

Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains
- PMID: 22120008
- PMCID: PMC4309644
- DOI: 10.1038/ng.969
Regions of focal DNA hypermethylation and long-range hypomethylation in colorectal cancer coincide with nuclear lamina-associated domains
Abstract
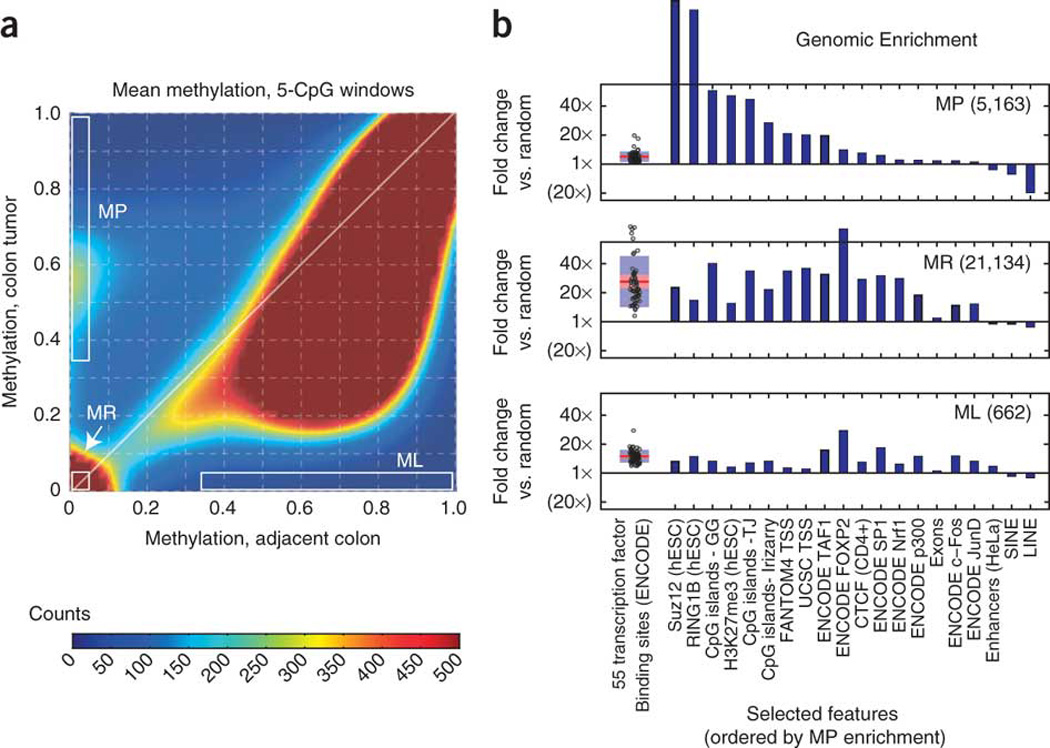
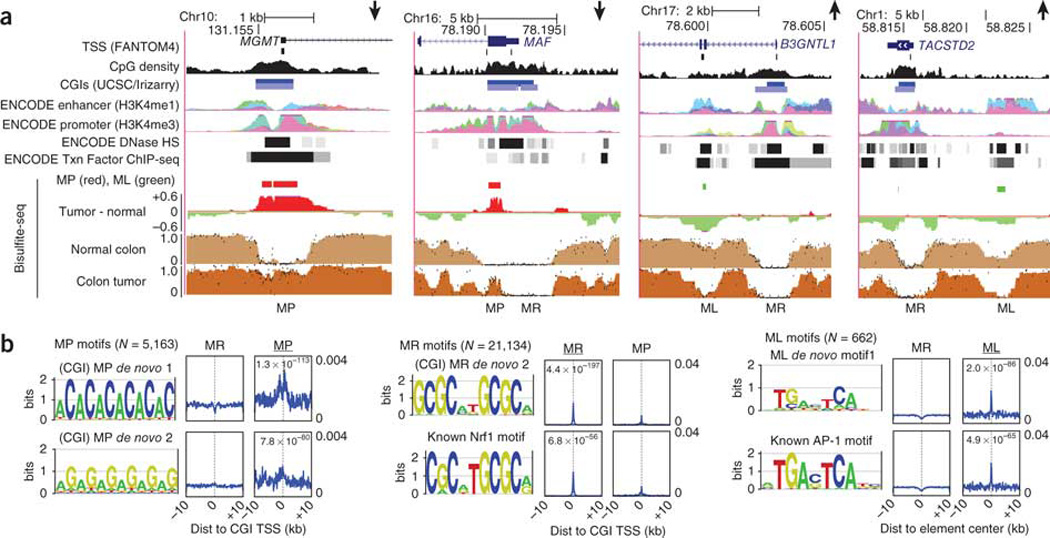
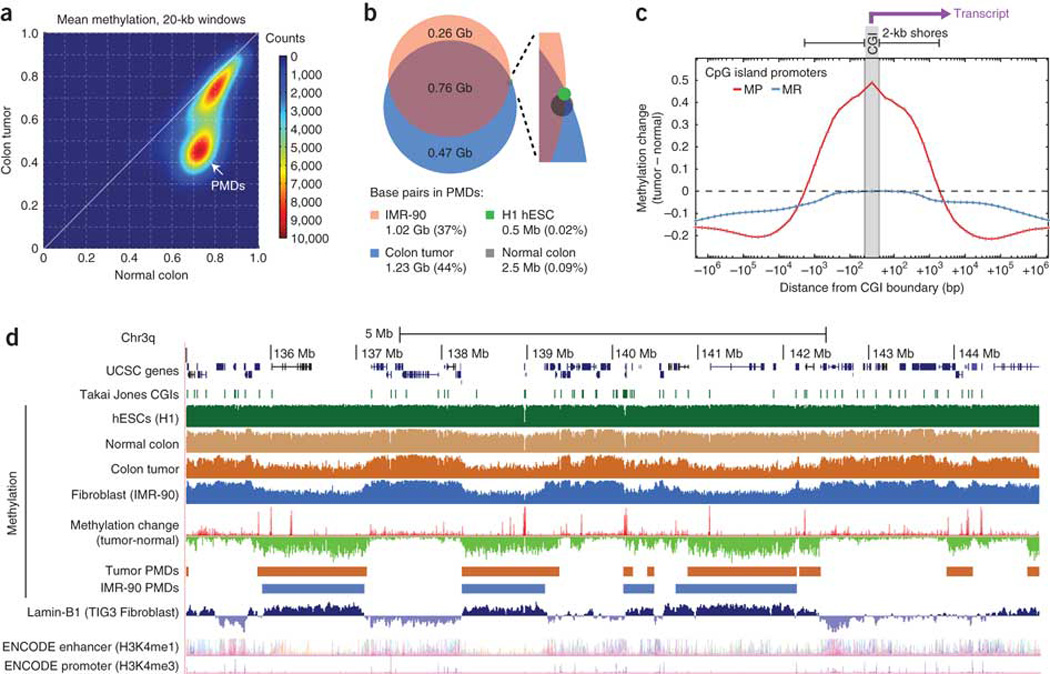
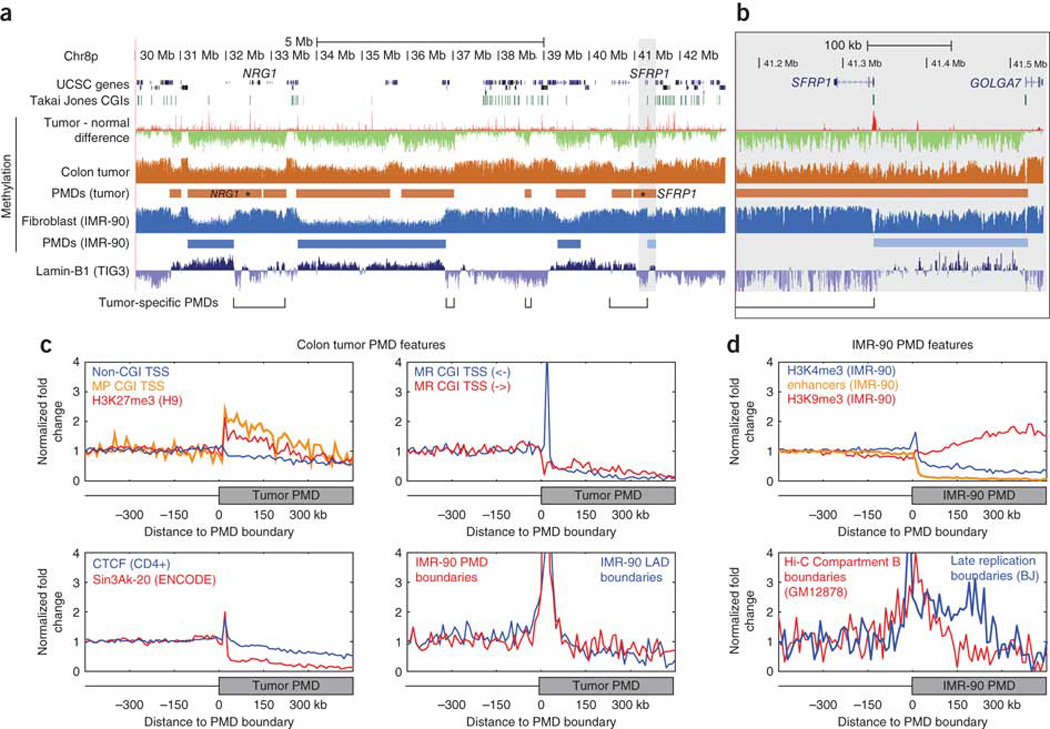
Extensive changes in DNA methylation are common in cancer and may contribute to oncogenesis through transcriptional silencing of tumor-suppressor genes. Genome-scale studies have yielded important insights into these changes but have focused on CpG islands or gene promoters. We used whole-genome bisulfite sequencing (bisulfite-seq) to comprehensively profile a primary human colorectal tumor and adjacent normal colon tissue at single-basepair resolution. Regions of focal hypermethylation in the tumor were located primarily at CpG islands and were concentrated within regions of long-range (>100 kb) hypomethylation. These hypomethylated domains covered nearly half of the genome and coincided with late replication and attachment to the nuclear lamina in human cell lines. We confirmed the confluence of hypermethylation and hypomethylation within these domains in 25 diverse colorectal tumors and matched adjacent tissue. We propose that widespread DNA methylation changes in cancer are linked to silencing programs orchestrated by the three-dimensional organization of chromatin within the nucleus.
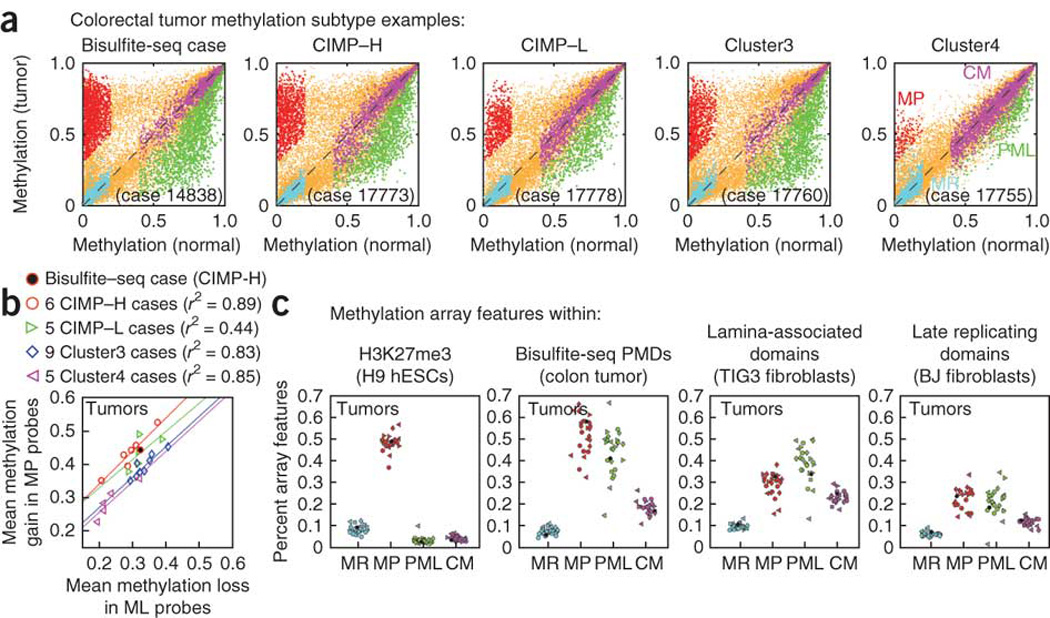
Figures

Similar articles
-
Non-CpG island promoter hypomethylation and miR-149 regulate the expression of SRPX2 in colorectal cancer.Int J Cancer. 2013 May 15;132(10):2303-15. doi: 10.1002/ijc.27921. Epub 2012 Nov 21. Int J Cancer. 2013. PMID: 23115050
-
Genome-wide methylation profiling identified novel differentially hypermethylated biomarker MPPED2 in colorectal cancer.Clin Epigenetics. 2019 Mar 7;11(1):41. doi: 10.1186/s13148-019-0628-y. Clin Epigenetics. 2019. PMID: 30846004 Free PMC article.
-
Susceptibility of nonpromoter CpG islands to de novo methylation in normal and neoplastic cells.J Natl Cancer Inst. 2001 Oct 3;93(19):1465-72. doi: 10.1093/jnci/93.19.1465. J Natl Cancer Inst. 2001. PMID: 11584062
-
MicroRNA Methylation in Colorectal Cancer.Adv Exp Med Biol. 2016;937:109-22. doi: 10.1007/978-3-319-42059-2_6. Adv Exp Med Biol. 2016. PMID: 27573897 Review.
-
[DNA methylation defects in sporadic and hereditary colorectal cancer].Gastroenterol Hepatol. 2012 Aug-Sep;35(7):480-7. doi: 10.1016/j.gastrohep.2012.01.010. Epub 2012 Mar 27. Gastroenterol Hepatol. 2012. PMID: 22459641 Review. Spanish.
Cited by
-
Causes of genome instability: the effect of low dose chemical exposures in modern society.Carcinogenesis. 2015 Jun;36 Suppl 1(Suppl 1):S61-88. doi: 10.1093/carcin/bgv031. Carcinogenesis. 2015. PMID: 26106144 Free PMC article. Review.
-
Large-Scale Topological Changes Restrain Malignant Progression in Colorectal Cancer.Cell. 2020 Sep 17;182(6):1474-1489.e23. doi: 10.1016/j.cell.2020.07.030. Epub 2020 Aug 24. Cell. 2020. PMID: 32841603 Free PMC article.
-
Interplay between the cancer genome and epigenome.Cell. 2013 Mar 28;153(1):38-55. doi: 10.1016/j.cell.2013.03.008. Cell. 2013. PMID: 23540689 Free PMC article. Review.
-
5-hydroxymethylcytosine marks promoters in colon that resist DNA hypermethylation in cancer.Genome Biol. 2015 Apr 1;16(1):69. doi: 10.1186/s13059-015-0605-5. Genome Biol. 2015. PMID: 25853800 Free PMC article.
-
The Crossroads of Cancer Epigenetics and Immune Checkpoint Therapy.Clin Cancer Res. 2023 Apr 3;29(7):1173-1182. doi: 10.1158/1078-0432.CCR-22-0784. Clin Cancer Res. 2023. PMID: 36449280 Free PMC article. Review.
References
-
- Jones PA, Baylin SB. The fundamental role of epigenetic events in cancer. Nat. Rev. Genet. 2002;3:415–428. - PubMed
-
- Gebhard C, et al. General transcription factor binding at CpG islands in normal cells correlates with resistance to de novo DNA methylation in cancer cells. Cancer Res. 2010;70:1398–1407. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
