

Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders
- PMID: 22090472
- PMCID: PMC3250079
- DOI: 10.1124/pr.110.003053
Therapeutic implications for striatal-enriched protein tyrosine phosphatase (STEP) in neuropsychiatric disorders
Abstract
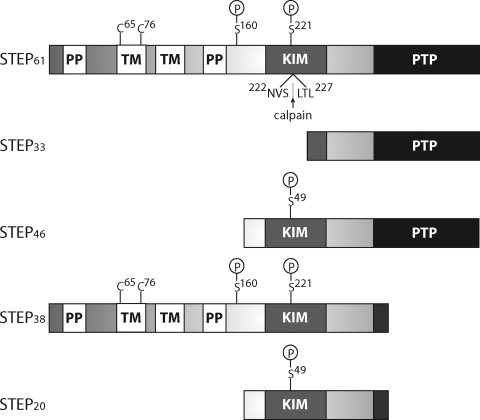
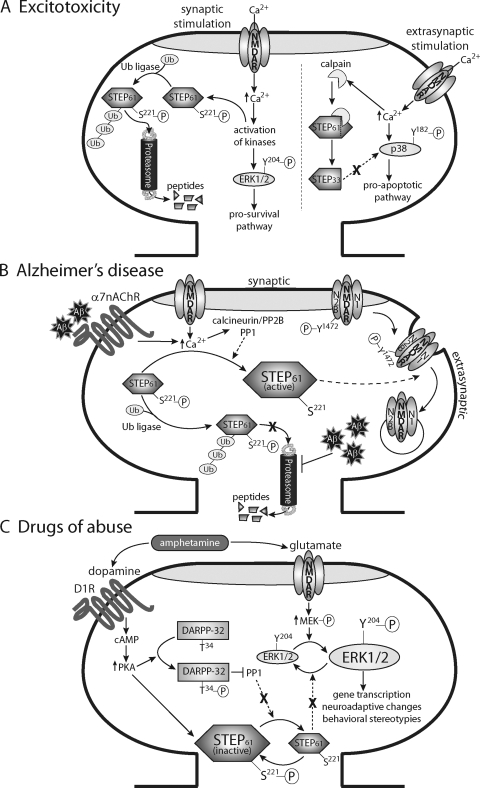
Striatal-enriched protein tyrosine phosphatase (STEP) is a brain-specific phosphatase that modulates key signaling molecules involved in synaptic plasticity and neuronal function. Targets include extracellular-regulated kinase 1 and 2 (ERK1/2), stress-activated protein kinase p38 (p38), the Src family tyrosine kinase Fyn, N-methyl-D-aspartate receptors (NMDARs), and α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs). STEP-mediated dephosphorylation of ERK1/2, p38, and Fyn leads to inactivation of these enzymes, whereas STEP-mediated dephosphorylation of surface NMDARs and AMPARs promotes their endocytosis. Accordingly, the current model of STEP function posits that it opposes long-term potentiation and promotes long-term depression. Phosphorylation, cleavage, dimerization, ubiquitination, and local translation all converge to maintain an appropriate balance of STEP in the central nervous system. Accumulating evidence over the past decade indicates that STEP dysregulation contributes to the pathophysiology of several neuropsychiatric disorders, including Alzheimer's disease, schizophrenia, fragile X syndrome, epileptogenesis, alcohol-induced memory loss, Huntington's disease, drug abuse, stroke/ischemia, and inflammatory pain. This comprehensive review discusses STEP expression and regulation and highlights how disrupted STEP function contributes to the pathophysiology of diverse neuropsychiatric disorders.
Figures



Similar articles
-
A common STEP in the synaptic pathology of diverse neuropsychiatric disorders.Yale J Biol Med. 2012 Dec;85(4):481-90. Epub 2012 Dec 13. Yale J Biol Med. 2012. PMID: 23239949 Free PMC article. Review.
-
Striatal-enriched Tyrosine Protein Phosphatase (STEP) in the Mechanisms of Depressive Disorders.Curr Protein Pept Sci. 2017 Aug 30;18(11):1152-1162. doi: 10.2174/1389203718666170710121532. Curr Protein Pept Sci. 2017. PMID: 28699511 Review.
-
Striatal-enriched protein tyrosine phosphatase (STEP) knockout mice have enhanced hippocampal memory.Eur J Neurosci. 2011 Jun;33(12):2288-98. doi: 10.1111/j.1460-9568.2011.07687.x. Epub 2011 Apr 19. Eur J Neurosci. 2011. PMID: 21501258 Free PMC article.
-
Disruption of striatal-enriched protein tyrosine phosphatase (STEP) function in neuropsychiatric disorders.Neurosci Res. 2014 Dec;89:1-9. doi: 10.1016/j.neures.2014.08.018. Epub 2014 Sep 10. Neurosci Res. 2014. PMID: 25218562 Free PMC article. Review.
-
An emerging role of STriatal-Enriched protein tyrosine Phosphatase in hyperexcitability-associated brain disorders.Neurobiol Dis. 2024 Oct 1;200:106641. doi: 10.1016/j.nbd.2024.106641. Epub 2024 Aug 17. Neurobiol Dis. 2024. PMID: 39159894 Review.
Cited by
-
The pathophysiology of fragile X (and what it teaches us about synapses).Annu Rev Neurosci. 2012;35:417-43. doi: 10.1146/annurev-neuro-060909-153138. Epub 2012 Apr 5. Annu Rev Neurosci. 2012. PMID: 22483044 Free PMC article. Review.
-
A common STEP in the synaptic pathology of diverse neuropsychiatric disorders.Yale J Biol Med. 2012 Dec;85(4):481-90. Epub 2012 Dec 13. Yale J Biol Med. 2012. PMID: 23239949 Free PMC article. Review.
-
Genetic manipulation of STEP reverses behavioral abnormalities in a fragile X syndrome mouse model.Genes Brain Behav. 2012 Jul;11(5):586-600. doi: 10.1111/j.1601-183X.2012.00781.x. Epub 2012 Apr 6. Genes Brain Behav. 2012. PMID: 22405502 Free PMC article.
-
Insight into the Role of the STriatal-Enriched Protein Tyrosine Phosphatase (STEP) in A2A Receptor-Mediated Effects in the Central Nervous System.Front Pharmacol. 2021 Apr 19;12:647742. doi: 10.3389/fphar.2021.647742. eCollection 2021. Front Pharmacol. 2021. PMID: 33953681 Free PMC article.
-
Cocaine-induced changes of synaptic transmission in the striatum are modulated by adenosine A2A receptors and involve the tyrosine phosphatase STEP.Neuropsychopharmacology. 2014 Feb;39(3):569-78. doi: 10.1038/npp.2013.229. Epub 2013 Aug 30. Neuropsychopharmacology. 2014. PMID: 23989619 Free PMC article.
References
-
- Adachi M, Sekiya M, Arimura Y, Takekawa M, Itoh F, Hinoda Y, Imai K, Yachi A. (1992) Protein-tyrosine phosphatase expression in pre-B cell NALM-6. Cancer Res 52:737–740 - PubMed
-
- Alvestad RM, Grosshans DR, Coultrap SJ, Nakazawa T, Yamamoto T, Browning MD. (2003) Tyrosine dephosphorylation and ethanol inhibition of N-methyl-d-aspartate receptor function. J Biol Chem 278:11020–11025 - PubMed
-
- Amaro S, Chamorro Á. (2011) Translational stroke research of the combination of thrombolysis and antioxidant therapy. Stroke 42:1495–1499 - PubMed
-
- Antar LN, Bassell GJ. (2003) Sunrise at the synapse: the FMRP mRNP shaping the synaptic interface. Neuron 37:555–558 - PubMed
-
- Antoniou X, Falconi M, Di Marino D, Borsello T. (2011) JNK3 as a therapeutic target for neurodegenerative diseases. J Alzheimers Dis 24:633–642 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous