

High-resolution analysis of intrahost genetic diversity in dengue virus serotype 1 infection identifies mixed infections
- PMID: 22090119
- PMCID: PMC3255838
- DOI: 10.1128/JVI.05985-11
High-resolution analysis of intrahost genetic diversity in dengue virus serotype 1 infection identifies mixed infections
Abstract
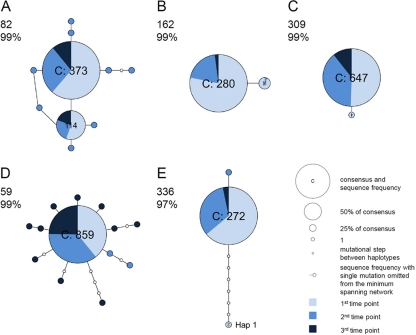
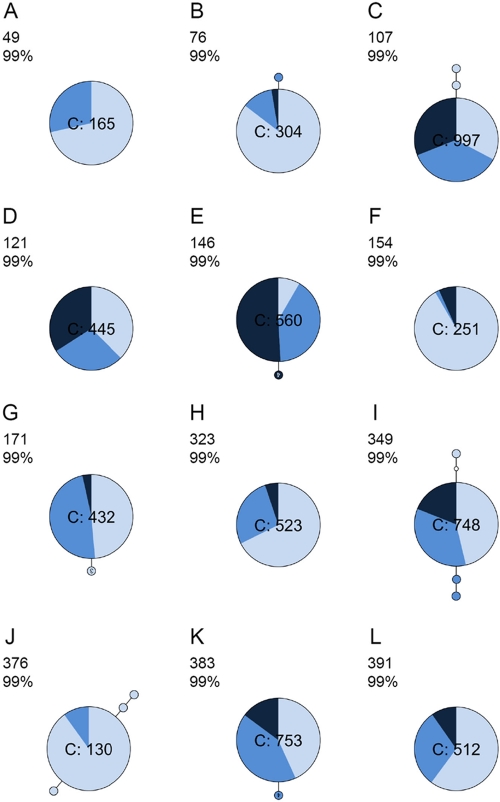
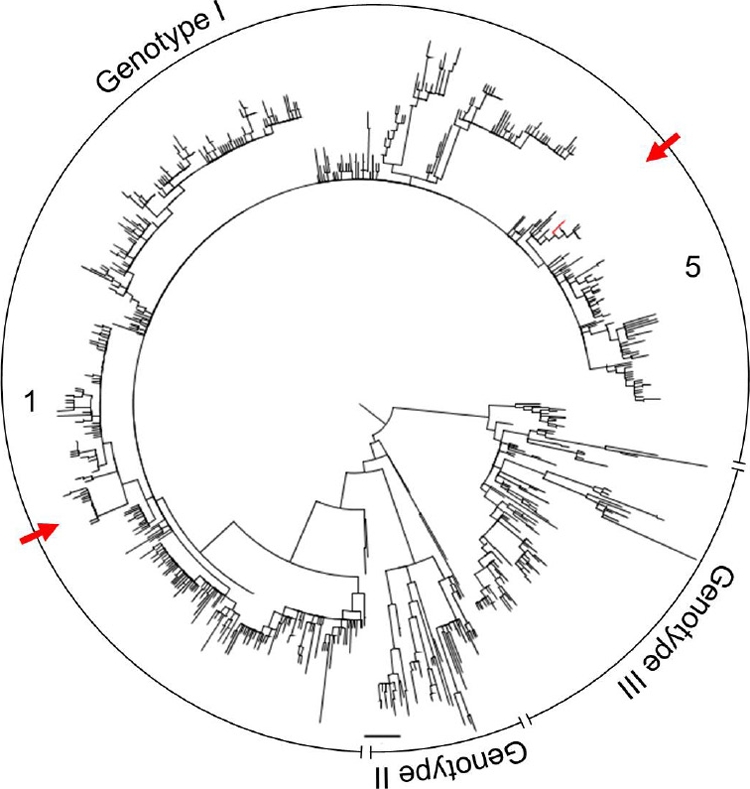
Little is known about the rate at which genetic variation is generated within intrahost populations of dengue virus (DENV) and what implications this diversity has for dengue pathogenesis, disease severity, and host immunity. Previous studies of intrahost DENV variation have used a low frequency of sampling and/or experimental methods that do not fully account for errors generated through amplification and sequencing of viral RNAs. We investigated the extent and pattern of genetic diversity in sequence data in domain III (DIII) of the envelope (E) gene in serial plasma samples (n = 49) taken from 17 patients infected with DENV type 1 (DENV-1), totaling some 8,458 clones. Statistically rigorous approaches were employed to account for artifactual variants resulting from amplification and sequencing, which we suggest have played a major role in previous studies of intrahost genetic variation. Accordingly, nucleotide sequence diversities of viral populations were very low, with conservative estimates of the average levels of genetic diversity ranging from 0 to 0.0013. Despite such sequence conservation, we observed clear evidence for mixed infection, with the presence of multiple phylogenetically distinct lineages present within the same host, while the presence of stop codon mutations in some samples suggests the action of complementation. In contrast to some previous studies we observed no relationship between the extent and pattern of DENV-1 genetic diversity and disease severity, immune status, or level of viremia.
Figures
Similar articles
-
Genome-wide patterns of intrahuman dengue virus diversity reveal associations with viral phylogenetic clade and interhost diversity.J Virol. 2012 Aug;86(16):8546-58. doi: 10.1128/JVI.00736-12. Epub 2012 May 30. J Virol. 2012. PMID: 22647702 Free PMC article.
-
Dengue Virus Serotype 2 Intrahost Diversity in Patients with Different Clinical Outcomes.Viruses. 2021 Feb 23;13(2):349. doi: 10.3390/v13020349. Viruses. 2021. PMID: 33672226 Free PMC article.
-
Increasing Clinical Severity during a Dengue Virus Type 3 Cuban Epidemic: Deep Sequencing of Evolving Viral Populations.J Virol. 2016 Apr 14;90(9):4320-4333. doi: 10.1128/JVI.02647-15. Print 2016 May. J Virol. 2016. PMID: 26889031 Free PMC article.
-
In Silico Analysis of Dengue Virus Serotype 2 Mutations Detected at the Intrahost Level in Patients with Different Clinical Outcomes.Microbiol Spectr. 2021 Oct 31;9(2):e0025621. doi: 10.1128/Spectrum.00256-21. Epub 2021 Sep 1. Microbiol Spectr. 2021. PMID: 34468189 Free PMC article.
-
Intrahost Selection Pressures Drive Rapid Dengue Virus Microevolution in Acute Human Infections.Cell Host Microbe. 2017 Sep 13;22(3):400-410.e5. doi: 10.1016/j.chom.2017.08.003. Cell Host Microbe. 2017. PMID: 28910637 Free PMC article.
Cited by
-
Single-reaction, multiplex, real-time rt-PCR for the detection, quantitation, and serotyping of dengue viruses.PLoS Negl Trop Dis. 2013 Apr 18;7(4):e2116. doi: 10.1371/journal.pntd.0002116. Print 2013. PLoS Negl Trop Dis. 2013. PMID: 23638191 Free PMC article.
-
Genetic diversity of the enteroviruses detected from cerebrospinal fluid (CSF) samples of patients with suspected aseptic meningitis in northern West Bank, Palestine in 2017.PLoS One. 2018 Dec 10;13(12):e0202243. doi: 10.1371/journal.pone.0202243. eCollection 2018. PLoS One. 2018. PMID: 30532168 Free PMC article. Clinical Trial.
-
Distribution of fitness in populations of dengue viruses.PLoS One. 2014 Sep 15;9(9):e107264. doi: 10.1371/journal.pone.0107264. eCollection 2014. PLoS One. 2014. PMID: 25222471 Free PMC article.
-
De novo assembly of highly diverse viral populations.BMC Genomics. 2012 Sep 13;13:475. doi: 10.1186/1471-2164-13-475. BMC Genomics. 2012. PMID: 22974120 Free PMC article.
-
Within-Host Viral Diversity: A Window into Viral Evolution.Annu Rev Virol. 2020 Sep 29;7(1):63-81. doi: 10.1146/annurev-virology-010320-061642. Epub 2020 Jun 8. Annu Rev Virol. 2020. PMID: 32511081 Free PMC article. Review.
References
-
- Aaskov J, Buzacott K, Thu HM, Lowry K, Holmes EC. 2006. Long-term transmission of defective RNA viruses in humans and Aedes mosquitoes. Science 311:236–238 - PubMed
-
- Altshuler D, et al. 2000. An SNP map of the human genome generated by reduced representation shotgun sequencing. Nature 407:513–516 - PubMed
-
- Clement M, Posada D, Crandall KA. 2000. TCS: a computer program to estimate gene genealogies. Mol. Ecol. 9:1657–1659 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
