

Protein misdirection inside and outside motor neurons in Amyotrophic Lateral Sclerosis (ALS): a possible clue for therapeutic strategies
- PMID: 22072931
- PMCID: PMC3211022
- DOI: 10.3390/ijms12106980
Protein misdirection inside and outside motor neurons in Amyotrophic Lateral Sclerosis (ALS): a possible clue for therapeutic strategies
Abstract
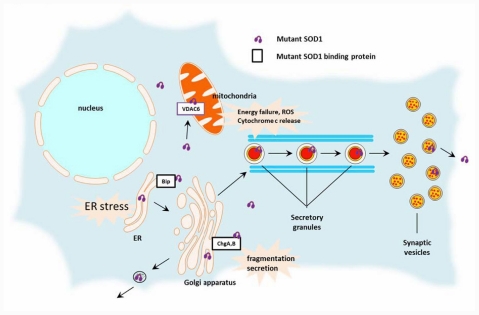
Amyotrophic lateral sclerosis (ALS) is a devastating neurodegenerative disease characterized by progressive muscle wasting and weakness with no effective cure. Emerging evidence supports the notion that the abnormal conformations of ALS-linked proteins play a central role in triggering the motor neuron degeneration. In particular, mutant types of superoxide dismutase 1 (SOD1) and TAR DNA binding protein 43kDa (TDP-43) are key molecules involved in the pathogenesis of familial and sporadic ALS, respectively. The commonalities of the two proteins include a propensity to aggregate and acquire detrimental conformations through oligomerization, fragmentation, or post-translational modification that may drive abnormal subcellular localizations. Although SOD1 is a major cytosolic protein, mutated SOD1 has been localized to mitochondria, endoplasmic reticulum, and even the extracellular space. The nuclear exclusion of TDP-43 is a pathological hallmark for ALS, although the pathogenic priority remains elusive. Nevertheless, these abnormal behaviors based on the protein misfolding are believed to induce diverse intracellular and extracellular events that may be tightly linked to non-cell-autonomous motor neuron death. The generation of mutant- or misfolded protein-specific antibodies would help to uncover the distribution and propagation of the ALS-linked proteins, and to design a therapeutic strategy to clear such species. Herein we review the literature regarding the mislocalization of ALS-linked proteins, especially mutant SOD1 and TDP-43 species, and discuss the rationale of molecular targeting strategies including immunotherapy.
Keywords: SOD1; TDP-43; antibody; non-cell-autonomous motor neuron death; seeding; subcellular localization.
Figures
Similar articles
-
Co-deposition of SOD1, TDP-43 and p62 proteinopathies in ALS: evidence for multifaceted pathways underlying neurodegeneration.Acta Neuropathol Commun. 2022 Aug 25;10(1):122. doi: 10.1186/s40478-022-01421-9. Acta Neuropathol Commun. 2022. PMID: 36008843 Free PMC article.
-
Pathological Modification of TDP-43 in Amyotrophic Lateral Sclerosis with SOD1 Mutations.Mol Neurobiol. 2019 Mar;56(3):2007-2021. doi: 10.1007/s12035-018-1218-2. Epub 2018 Jul 7. Mol Neurobiol. 2019. PMID: 29982983 Free PMC article.
-
Aberrant localization of FUS and TDP43 is associated with misfolding of SOD1 in amyotrophic lateral sclerosis.PLoS One. 2012;7(4):e35050. doi: 10.1371/journal.pone.0035050. Epub 2012 Apr 6. PLoS One. 2012. PMID: 22493728 Free PMC article.
-
[Implications of successful immunotherapy in ALS model mice].Brain Nerve. 2008 Jun;60(6):643-51. Brain Nerve. 2008. PMID: 18567360 Review. Japanese.
-
Does wild-type Cu/Zn-superoxide dismutase have pathogenic roles in amyotrophic lateral sclerosis?Transl Neurodegener. 2020 Aug 19;9(1):33. doi: 10.1186/s40035-020-00209-y. Transl Neurodegener. 2020. PMID: 32811540 Free PMC article. Review.
Cited by
-
Exposure to environmental toxicants and pathogenesis of amyotrophic lateral sclerosis: state of the art and research perspectives.Int J Mol Sci. 2013 Jul 24;14(8):15286-311. doi: 10.3390/ijms140815286. Int J Mol Sci. 2013. PMID: 23887652 Free PMC article. Review.
-
Golgi Fragmentation in Neurodegenerative Diseases: Is There a Common Cause?Cells. 2019 Jul 19;8(7):748. doi: 10.3390/cells8070748. Cells. 2019. PMID: 31331075 Free PMC article. Review.
-
Evidence for prion-like mechanisms in several neurodegenerative diseases: potential implications for immunotherapy.Clin Dev Immunol. 2013;2013:473706. doi: 10.1155/2013/473706. Epub 2013 Oct 20. Clin Dev Immunol. 2013. PMID: 24228054 Free PMC article. Review.
-
An Allosteric Pathway in Copper, Zinc Superoxide Dismutase Unravels the Molecular Mechanism of the G93A Amyotrophic Lateral Sclerosis-Linked Mutation.J Phys Chem Lett. 2019 Dec 19;10(24):7740-7744. doi: 10.1021/acs.jpclett.9b02868. Epub 2019 Dec 3. J Phys Chem Lett. 2019. PMID: 31747286 Free PMC article.
-
Immunotherapies for Neurodegenerative Diseases.Front Neurol. 2021 Jun 7;12:654739. doi: 10.3389/fneur.2021.654739. eCollection 2021. Front Neurol. 2021. PMID: 34163421 Free PMC article. Review.
References
-
- Kabashi E, Durham HD. Failure of protein quality control in amyotrophic lateral sclerosis. Biochim. Biophys. Acta. 2006;1762:1038–1050. - PubMed
-
- Hart PJ. Pathogenic superoxide dismutase structure, folding, aggregation and turnover. Curr. Opin. Chem. Biol. 2006;10:131–138. - PubMed
-
- Ticozzi N, Ratti A, Silani V. Protein aggregation and defective RNA metabolism as mechanisms for motor neuron damage. CNS Neurol. Disord. Drug Targets. 2010;9:285–296. - PubMed
-
- Takahashi K, Nakamura H, Okada E. Hereditary amyotrophic lateral sclerosis. Histochemical and electron microscopic study of hyaline inclusions in motor neurons. Arch. Neurol. 1972;27:292–299. - PubMed
-
- Sun CN, Araoz C, Lucas G, Morgan PN, White HJ. Amyotrophic lateral sclerosis. Inclusion bodies in a case of the classic sporadic form. Ann. Clin. Lab. Sci. 1975;5:38–44. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
