

A yeast functional screen predicts new candidate ALS disease genes
- PMID: 22065782
- PMCID: PMC3248518
- DOI: 10.1073/pnas.1109434108
A yeast functional screen predicts new candidate ALS disease genes
Erratum in
-
Correction for Couthouis et al., A yeast functional screen predicts new candidate ALS disease genes.Proc Natl Acad Sci U S A. 2023 Jan 3;120(1):e2220845120. doi: 10.1073/pnas.2220845120. Epub 2022 Dec 28. Proc Natl Acad Sci U S A. 2023. PMID: 36577079 Free PMC article. No abstract available.
Abstract
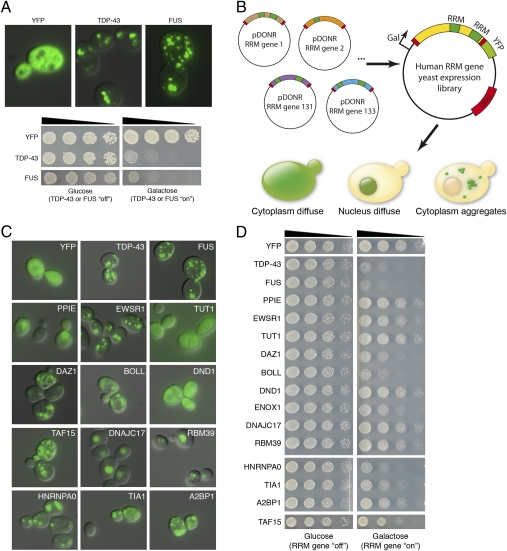
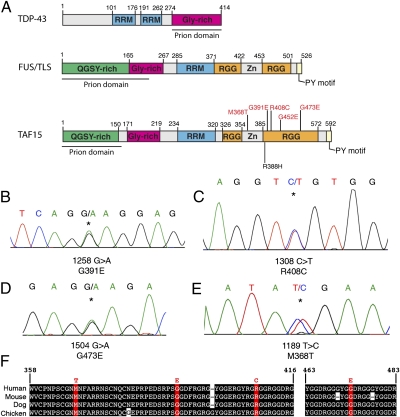
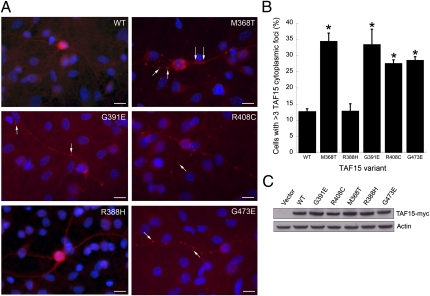
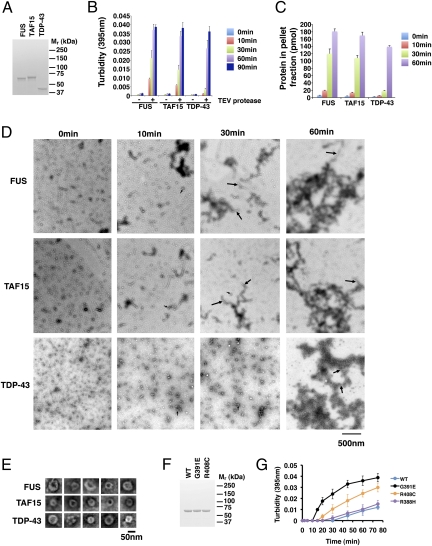
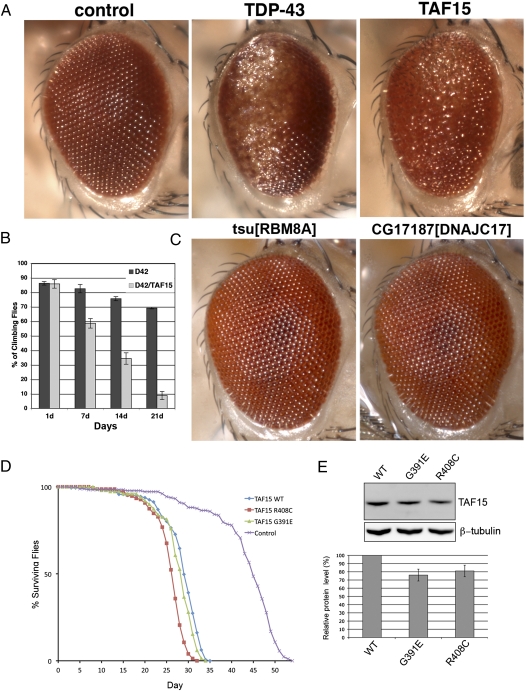
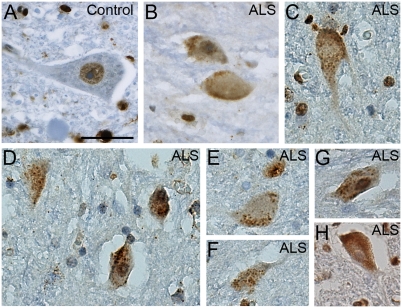
Amyotrophic lateral sclerosis (ALS) is a devastating and universally fatal neurodegenerative disease. Mutations in two related RNA-binding proteins, TDP-43 and FUS, that harbor prion-like domains, cause some forms of ALS. There are at least 213 human proteins harboring RNA recognition motifs, including FUS and TDP-43, raising the possibility that additional RNA-binding proteins might contribute to ALS pathogenesis. We performed a systematic survey of these proteins to find additional candidates similar to TDP-43 and FUS, followed by bioinformatics to predict prion-like domains in a subset of them. We sequenced one of these genes, TAF15, in patients with ALS and identified missense variants, which were absent in a large number of healthy controls. These disease-associated variants of TAF15 caused formation of cytoplasmic foci when expressed in primary cultures of spinal cord neurons. Very similar to TDP-43 and FUS, TAF15 aggregated in vitro and conferred neurodegeneration in Drosophila, with the ALS-linked variants having a more severe effect than wild type. Immunohistochemistry of postmortem spinal cord tissue revealed mislocalization of TAF15 in motor neurons of patients with ALS. We propose that aggregation-prone RNA-binding proteins might contribute very broadly to ALS pathogenesis and the genes identified in our yeast functional screen, coupled with prion-like domain prediction analysis, now provide a powerful resource to facilitate ALS disease gene discovery.
Conflict of interest statement
Conflict of interest statement: A.D.G. is an inventor on patents and patent applications that have been licensed to FoldRx Pharmaceuticals.
Figures
Comment in
-
Motor neuron disease: Functional screening identifies novel candidate risk genes in amyotrophic lateral sclerosis.Nat Rev Neurol. 2011 Dec 6;8(1):1. doi: 10.1038/nrneurol.2011.203. Nat Rev Neurol. 2011. PMID: 22143366 No abstract available.
Similar articles
-
Evaluating the role of the FUS/TLS-related gene EWSR1 in amyotrophic lateral sclerosis.Hum Mol Genet. 2012 Jul 1;21(13):2899-911. doi: 10.1093/hmg/dds116. Epub 2012 Mar 27. Hum Mol Genet. 2012. PMID: 22454397 Free PMC article.
-
Distinct and shared functions of ALS-associated proteins TDP-43, FUS and TAF15 revealed by multisystem analyses.Nat Commun. 2016 Jul 5;7:12143. doi: 10.1038/ncomms12143. Nat Commun. 2016. PMID: 27378374 Free PMC article.
-
The tip of the iceberg: RNA-binding proteins with prion-like domains in neurodegenerative disease.Brain Res. 2012 Jun 26;1462:61-80. doi: 10.1016/j.brainres.2012.01.016. Epub 2012 Jan 21. Brain Res. 2012. PMID: 22445064 Free PMC article. Review.
-
RNA-binding ability of FUS regulates neurodegeneration, cytoplasmic mislocalization and incorporation into stress granules associated with FUS carrying ALS-linked mutations.Hum Mol Genet. 2013 Mar 15;22(6):1193-205. doi: 10.1093/hmg/dds526. Epub 2012 Dec 20. Hum Mol Genet. 2013. PMID: 23257289 Free PMC article.
-
RNA-binding proteins with prion-like domains in ALS and FTLD-U.Prion. 2011 Jul-Sep;5(3):179-87. doi: 10.4161/pri.5.3.17230. Epub 2011 Jul 1. Prion. 2011. PMID: 21847013 Free PMC article. Review.
Cited by
-
FUS/TLS acts as an aggregation-dependent modifier of polyglutamine disease model mice.Sci Rep. 2016 Oct 14;6:35236. doi: 10.1038/srep35236. Sci Rep. 2016. PMID: 27739513 Free PMC article.
-
DDX17 is involved in DNA damage repair and modifies FUS toxicity in an RGG-domain dependent manner.Acta Neuropathol. 2021 Sep;142(3):515-536. doi: 10.1007/s00401-021-02333-z. Epub 2021 Jun 1. Acta Neuropathol. 2021. PMID: 34061233 Free PMC article.
-
Linking RNA Dysfunction and Neurodegeneration in Amyotrophic Lateral Sclerosis.Neurotherapeutics. 2015 Apr;12(2):340-51. doi: 10.1007/s13311-015-0340-3. Neurotherapeutics. 2015. PMID: 25689976 Free PMC article. Review.
-
Quantitative analysis of the detergent-insoluble brain proteome in frontotemporal lobar degeneration using SILAC internal standards.J Proteome Res. 2012 May 4;11(5):2721-38. doi: 10.1021/pr2010814. Epub 2012 Apr 4. J Proteome Res. 2012. PMID: 22416763 Free PMC article.
-
Pathological mechanisms underlying TDP-43 driven neurodegeneration in FTLD-ALS spectrum disorders.Hum Mol Genet. 2013 Oct 15;22(R1):R77-87. doi: 10.1093/hmg/ddt349. Epub 2013 Jul 29. Hum Mol Genet. 2013. PMID: 23900071 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
- NS072561/NS/NINDS NIH HHS/United States
- T32 AG000255/AG/NIA NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- T32-AG00255/AG/NIA NIH HHS/United States
- AG17586/AG/NIA NIH HHS/United States
- R01NS065782/NS/NINDS NIH HHS/United States
- 5R21NS067354-02/NS/NINDS NIH HHS/United States
- DP2 OD002177/OD/NIH HHS/United States
- R01 NS065782/NS/NINDS NIH HHS/United States
- R01 NS065317/NS/NINDS NIH HHS/United States
- R21 NS072561/NS/NINDS NIH HHS/United States
- R21 NS067354/NS/NINDS NIH HHS/United States
- R01 AG26251-03A1/AG/NIA NIH HHS/United States
- P50 AG16574/AG/NIA NIH HHS/United States
- P01 AG032953/AG/NIA NIH HHS/United States
- DP2 OD004417/OD/NIH HHS/United States
- P30 AG010124/AG/NIA NIH HHS/United States
- 1R01NS065317/NS/NINDS NIH HHS/United States
- P01 AG009215/AG/NIA NIH HHS/United States
- AG10124/AG/NIA NIH HHS/United States
- U24 AG021886/AG/NIA NIH HHS/United States
- P01-AG-09215/AG/NIA NIH HHS/United States
- P50 AG016574/AG/NIA NIH HHS/United States
- P01 AG017586/AG/NIA NIH HHS/United States
- 1DP2OD002177-01/OD/NIH HHS/United States
- 1DP2OD004417/OD/NIH HHS/United States
- R01 AG026251/AG/NIA NIH HHS/United States
- U24 AG21886/AG/NIA NIH HHS/United States
- R01 NS056070/NS/NINDS NIH HHS/United States
- NS056070/NS/NINDS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
