

L-DOPA-Induced Dyskinesia and Abnormal Signaling in Striatal Medium Spiny Neurons: Focus on Dopamine D1 Receptor-Mediated Transmission
- PMID: 22028687
- PMCID: PMC3199545
- DOI: 10.3389/fnbeh.2011.00071
L-DOPA-Induced Dyskinesia and Abnormal Signaling in Striatal Medium Spiny Neurons: Focus on Dopamine D1 Receptor-Mediated Transmission
Abstract
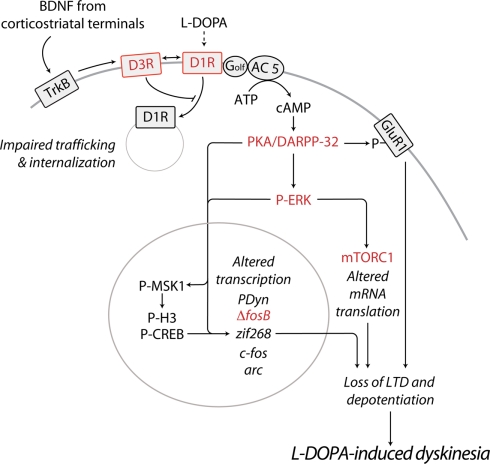
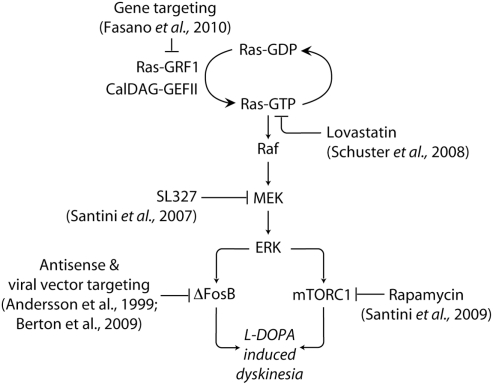
Dyskinesia is a serious motor complication caused by prolonged administration of l-DOPA to patients affected by Parkinson's disease. Accumulating evidence indicates that l-DOPA-induced dyskinesia (LID) is primarily caused by the development of sensitized dopamine D1 receptor (D1R) transmission in the medium spiny neurons (MSNs) of the striatum. This phenomenon, combined with chronic administration of l-DOPA, leads to persistent and intermittent hyper-activation of the cAMP signaling cascade. Activation of cAMP signaling results in increased activity of the cAMP-dependent protein kinase (PKA) and of the dopamine- and cAMP-dependent phosphoprotein of 32 kDa (DARPP-32), which regulate several downstream effector targets implicated in the control of the excitability of striatal MSNs. Dyskinesia is also accompanied by augmented activity of the extracellular signal-regulated kinases (ERK) and the mammalian target of rapamycin complex 1 (mTORC1), which are involved in the control of transcriptional and translational efficiency. Pharmacological or genetic interventions aimed at reducing abnormal signal transduction at the level of these various intracellular cascades have been shown to attenuate LID in different animal models. For instance, LID is reduced in mice deficient for DARPP-32, or following inhibition of PKA. Blockade of ERK obtained genetically or using specific inhibitors is also able to attenuate dyskinetic behavior in rodents and non-human primates. Finally, administration of rapamycin, a drug which blocks mTORC1, results in a strong reduction of LID. This review focuses on the abnormalities in signaling affecting the D1R-expressing MSNs and on their potential relevance for the design of novel anti-dyskinetic therapies.
Keywords: Parkinson’s disease; cAMP; dopamine- and cAMP-regulated phosphoprotein 32 kDa; extracellular signal-regulated protein kinases; immediate early genes; mammalian target of rapamycin.
Figures
Similar articles
-
Dopamine- and cAMP-regulated phosphoprotein of 32-kDa (DARPP-32)-dependent activation of extracellular signal-regulated kinase (ERK) and mammalian target of rapamycin complex 1 (mTORC1) signaling in experimental parkinsonism.J Biol Chem. 2012 Aug 10;287(33):27806-12. doi: 10.1074/jbc.M112.388413. Epub 2012 Jun 29. J Biol Chem. 2012. PMID: 22753408 Free PMC article.
-
Rapamycin, by Inhibiting mTORC1 Signaling, Prevents the Loss of Striatal Bidirectional Synaptic Plasticity in a Rat Model of L-DOPA-Induced Dyskinesia.Front Aging Neurosci. 2020 Aug 11;12:230. doi: 10.3389/fnagi.2020.00230. eCollection 2020. Front Aging Neurosci. 2020. PMID: 32848709 Free PMC article.
-
Critical involvement of cAMP/DARPP-32 and extracellular signal-regulated protein kinase signaling in L-DOPA-induced dyskinesia.J Neurosci. 2007 Jun 27;27(26):6995-7005. doi: 10.1523/JNEUROSCI.0852-07.2007. J Neurosci. 2007. PMID: 17596448 Free PMC article.
-
Signal transduction in L-DOPA-induced dyskinesia: from receptor sensitization to abnormal gene expression.J Neural Transm (Vienna). 2018 Aug;125(8):1171-1186. doi: 10.1007/s00702-018-1847-7. Epub 2018 Feb 2. J Neural Transm (Vienna). 2018. PMID: 29396608 Free PMC article. Review.
-
Potential for targeting dopamine/DARPP-32 signaling in neuropsychiatric and neurodegenerative disorders.Expert Opin Ther Targets. 2017 Mar;21(3):259-272. doi: 10.1080/14728222.2017.1279149. Epub 2017 Jan 13. Expert Opin Ther Targets. 2017. PMID: 28052701 Review.
Cited by
-
Effects of bromelain on motor responses following intra-medial forebrain bundle 6-OHDA injection in rat model of parkinsonism.Metab Brain Dis. 2019 Dec;34(6):1557-1564. doi: 10.1007/s11011-019-00462-9. Epub 2019 Jul 22. Metab Brain Dis. 2019. PMID: 31332728
-
5-Hydroxytryptophan Reduces Levodopa-Induced Dyskinesia via Regulating AKT/mTOR/S6K and CREB/ΔFosB Signals in a Mouse Model of Parkinson's Disease.Biomol Ther (Seoul). 2023 Jul 1;31(4):402-410. doi: 10.4062/biomolther.2022.141. Epub 2023 Mar 15. Biomol Ther (Seoul). 2023. PMID: 36918741 Free PMC article.
-
Integrated transcriptome expression profiling reveals a novel lncRNA associated with L-DOPA-induced dyskinesia in a rat model of Parkinson's disease.Aging (Albany NY). 2020 Jan 10;12(1):718-739. doi: 10.18632/aging.102652. Epub 2020 Jan 10. Aging (Albany NY). 2020. PMID: 31929116 Free PMC article.
-
Loss of Striatonigral GABAergic Presynaptic Inhibition Enables Motor Sensitization in Parkinsonian Mice.Neuron. 2015 Sep 2;87(5):976-88. doi: 10.1016/j.neuron.2015.08.022. Neuron. 2015. PMID: 26335644 Free PMC article.
-
Reduced striatal M4-cholinergic signaling following dopamine loss contributes to parkinsonian and l-DOPA-induced dyskinetic behaviors.Sci Adv. 2024 Nov 22;10(47):eadp6301. doi: 10.1126/sciadv.adp6301. Epub 2024 Nov 20. Sci Adv. 2024. PMID: 39565858 Free PMC article.
References
-
- Ahmed M. R., Berthet A., Bychkov E., Porras G., Li Q., Bioulac B. H., Carl Y. T., Bloch B., Kook S., Aubert I., Dovero S., Doudnikoff E., Gurevich V. V., Gurevich E. V., Bezard E. (2010). Lentiviral overexpression of GRK6 alleviates L-dopa-induced dyskinesia in experimental Parkinson’s disease. Sci. Transl. Med. 2, 28ra28.10.1126/scitranslmed.3000664 - DOI - PMC - PubMed
-
- Andersson M., Hilbertson A., Cenci M. A. (1999). Striatal fosB expression is causally linked with l-DOPA-induced abnormal involuntary movements and the associated upregulation of striatal prodynorphin mRNA in a rat model of Parkinson’s disease. Neurobiol. Dis. 6, 461–47410.1006/nbdi.1999.0259 - DOI - PubMed
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous