

Shift in brain metabolism in late onset Alzheimer's disease: implications for biomarkers and therapeutic interventions
- PMID: 22024249
- PMCID: PMC3658304
- DOI: 10.1016/j.mam.2011.10.005
Shift in brain metabolism in late onset Alzheimer's disease: implications for biomarkers and therapeutic interventions
Abstract
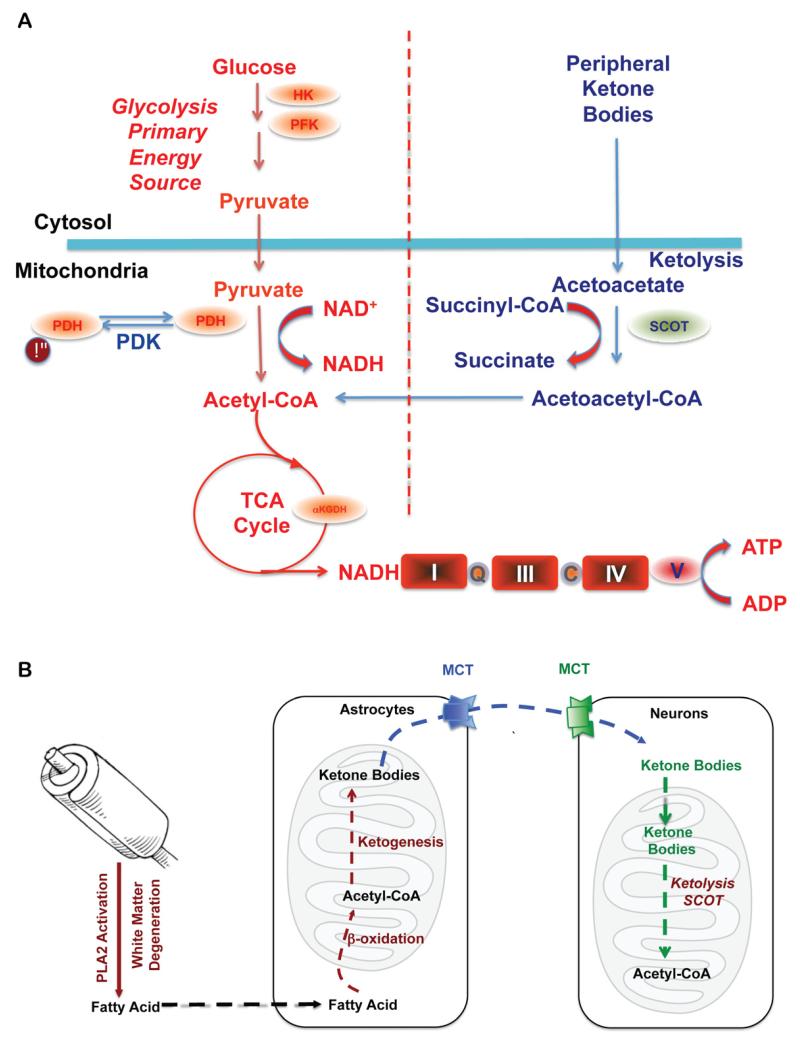
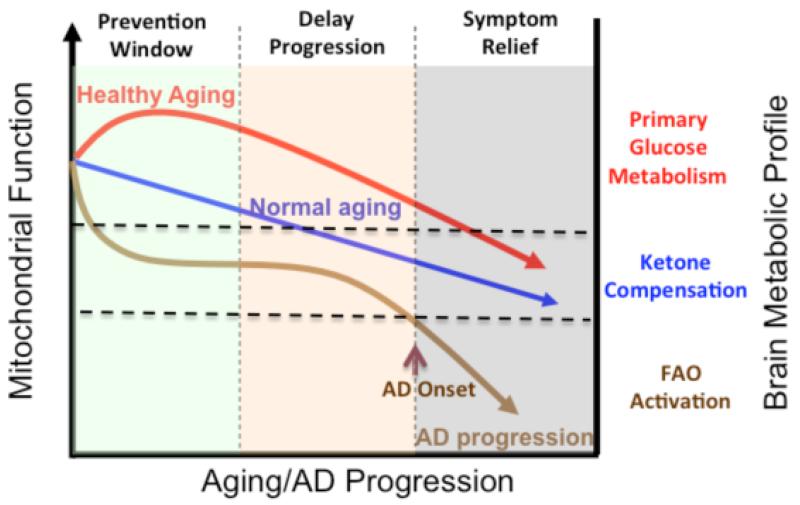
Alzheimer's is a neurodegenerative disease with a complex and progressive pathological phenotype characterized first by hypometabolism and impaired mitochondrial bioenergetics followed by pathological burden. Increasing evidence indicates an antecedent and potentially causal role of mitochondrial bioenergetic deficits and brain hypometabolism coupled with increased mitochondrial oxidative stress in AD pathogenesis. Compromised mitochondrial bioenergetics lead to over-production of and mitochondrial accumulation of β-amyloid, which is coupled with oxidative stress. Collectively, this results in a shift in brain metabolic profile from glucose-driven bioenergetics towards a compensatory, but less efficient, ketogenic pathway. We propose that the compensatory shift from a primarily aerobic glycolysis pathway to a ketogenic/fatty acid β-oxidation pathway eventually leads to white matter degeneration. The essential role of mitochondrial bioenergetics and the unique trajectory of compensatory metabolic adaptations in brain enable a bioenergetic-centric strategy for development of biomarkers. From a therapeutic perspective, this trajectory of alterations in brain metabolic capacity enables disease-stage specific strategies to target brain metabolism for disease prevention and treatment. A combination of nutraceutical and pharmaceutical interventions that enhance glucose-driven metabolic activity and potentiate mitochondrial bioenergetic function could prevent the antecedent decline in brain glucose metabolism, promote healthy aging and prevent AD. Alternatively, during the prodromal incipient phase of AD, sustained activation of ketogenic metabolic pathways coupled with supplementation of the alternative fuel source, ketone bodies, could sustain mitochondrial bioenergetic function to prevent or delay further progression of the disease.
Copyright © 2011 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Estrogen regulation of mitochondrial bioenergetics: implications for prevention of Alzheimer's disease.Adv Pharmacol. 2012;64:327-71. doi: 10.1016/B978-0-12-394816-8.00010-6. Adv Pharmacol. 2012. PMID: 22840752 Free PMC article. Review.
-
Targeting mitochondrial bioenergetics for Alzheimer's prevention and treatment.Curr Pharm Des. 2011;17(31):3474-9. doi: 10.2174/138161211798072517. Curr Pharm Des. 2011. PMID: 21902662 Free PMC article.
-
2-Deoxy-D-glucose treatment induces ketogenesis, sustains mitochondrial function, and reduces pathology in female mouse model of Alzheimer's disease.PLoS One. 2011;6(7):e21788. doi: 10.1371/journal.pone.0021788. Epub 2011 Jul 1. PLoS One. 2011. PMID: 21747957 Free PMC article.
-
White Matter Lipids as a Ketogenic Fuel Supply in Aging Female Brain: Implications for Alzheimer's Disease.EBioMedicine. 2015 Nov 3;2(12):1888-904. doi: 10.1016/j.ebiom.2015.11.002. eCollection 2015 Dec. EBioMedicine. 2015. PMID: 26844268 Free PMC article.
-
Targeting the prodromal stage of Alzheimer's disease: bioenergetic and mitochondrial opportunities.Neurotherapeutics. 2015 Jan;12(1):66-80. doi: 10.1007/s13311-014-0324-8. Neurotherapeutics. 2015. PMID: 25534394 Free PMC article. Review.
Cited by
-
Estrogen regulation of mitochondrial bioenergetics: implications for prevention of Alzheimer's disease.Adv Pharmacol. 2012;64:327-71. doi: 10.1016/B978-0-12-394816-8.00010-6. Adv Pharmacol. 2012. PMID: 22840752 Free PMC article. Review.
-
Intestinal endogenous metabolites affect neuroinflammation in 5×FAD mice by mediating "gut-brain" axis and the intervention with Chinese Medicine.Alzheimers Res Ther. 2024 Oct 14;16(1):222. doi: 10.1186/s13195-024-01587-5. Alzheimers Res Ther. 2024. PMID: 39396997 Free PMC article.
-
Triheptanoin Mitigates Brain ATP Depletion and Mitochondrial Dysfunction in a Mouse Model of Alzheimer's Disease.J Alzheimers Dis. 2020;78(1):425-437. doi: 10.3233/JAD-200594. J Alzheimers Dis. 2020. PMID: 33016909 Free PMC article.
-
Global Metabolic Shifts in Age and Alzheimer's Disease Mouse Brains Pivot at NAD+/NADH Redox Sites.J Alzheimers Dis. 2019;71(1):119-140. doi: 10.3233/JAD-190408. J Alzheimers Dis. 2019. PMID: 31356210 Free PMC article.
-
Ketogenic diet improves motor performance but not cognition in two mouse models of Alzheimer's pathology.PLoS One. 2013 Sep 12;8(9):e75713. doi: 10.1371/journal.pone.0075713. eCollection 2013. PLoS One. 2013. PMID: 24069439 Free PMC article.
References
-
- Consensus report of the Working Group “Molecular and Biochemical Markers of Alzheimer’s Disease”. The Ronald and Nancy Reagan Research Institute of the Alzheimer’s Association and the National Institute on Aging Working Group. Neurobiol Aging. 1998;19(2):109–116. - PubMed
-
- Association A.s. 2011 Alzheimer’s Disease. 2011 Facts and Figures 12.
-
- Atamna H, Frey WH. Mechanisms of mitochondrial dysfunction and energy deficiency in Alzheimer’s disease. Mitochondrion. (2nd) 2007 - PubMed
-
- Auestad N, Korsak RA, Morrow JW, Edmond J. Fatty acid oxidation and ketogenesis by astrocytes in primary culture. J Neurochem. 1991;56(4):1376–1386. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
