

Raw sewage harbors diverse viral populations
- PMID: 21972239
- PMCID: PMC3187576
- DOI: 10.1128/mBio.00180-11
Raw sewage harbors diverse viral populations
Abstract
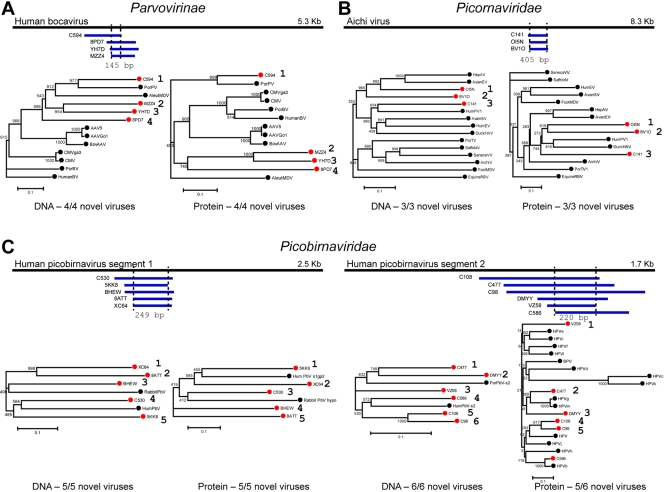
At this time, about 3,000 different viruses are recognized, but metagenomic studies suggest that these viruses are a small fraction of the viruses that exist in nature. We have explored viral diversity by deep sequencing nucleic acids obtained from virion populations enriched from raw sewage. We identified 234 known viruses, including 17 that infect humans. Plant, insect, and algal viruses as well as bacteriophages were also present. These viruses represented 26 taxonomic families and included viruses with single-stranded DNA (ssDNA), double-stranded DNA (dsDNA), positive-sense ssRNA [ssRNA(+)], and dsRNA genomes. Novel viruses that could be placed in specific taxa represented 51 different families, making untreated wastewater the most diverse viral metagenome (genetic material recovered directly from environmental samples) examined thus far. However, the vast majority of sequence reads bore little or no sequence relation to known viruses and thus could not be placed into specific taxa. These results show that the vast majority of the viruses on Earth have not yet been characterized. Untreated wastewater provides a rich matrix for identifying novel viruses and for studying virus diversity.
Importance: At this time, virology is focused on the study of a relatively small number of viral species. Specific viruses are studied either because they are easily propagated in the laboratory or because they are associated with disease. The lack of knowledge of the size and characteristics of the viral universe and the diversity of viral genomes is a roadblock to understanding important issues, such as the origin of emerging pathogens and the extent of gene exchange among viruses. Untreated wastewater is an ideal system for assessing viral diversity because virion populations from large numbers of individuals are deposited and because raw sewage itself provides a rich environment for the growth of diverse host species and thus their viruses. These studies suggest that the viral universe is far more vast and diverse than previously suspected.
Figures




Similar articles
-
Phytovirome Analysis of Wild Plant Populations: Comparison of Double-Stranded RNA and Virion-Associated Nucleic Acid Metagenomic Approaches.J Virol. 2019 Dec 12;94(1):e01462-19. doi: 10.1128/JVI.01462-19. Print 2019 Dec 12. J Virol. 2019. PMID: 31597769 Free PMC article.
-
Census of the viral metagenome within an activated sludge microbial assemblage.Appl Environ Microbiol. 2010 Apr;76(8):2673-7. doi: 10.1128/AEM.02520-09. Epub 2010 Feb 12. Appl Environ Microbiol. 2010. PMID: 20154108 Free PMC article.
-
High variety of known and new RNA and DNA viruses of diverse origins in untreated sewage.J Virol. 2012 Nov;86(22):12161-75. doi: 10.1128/JVI.00869-12. Epub 2012 Aug 29. J Virol. 2012. PMID: 22933275 Free PMC article.
-
RNA Viruses in Aquatic Ecosystems through the Lens of Ecological Genomics and Transcriptomics.Viruses. 2022 Mar 28;14(4):702. doi: 10.3390/v14040702. Viruses. 2022. PMID: 35458432 Free PMC article. Review.
-
Virus classification - where do you draw the line?Arch Virol. 2018 Aug;163(8):2037-2046. doi: 10.1007/s00705-018-3938-z. Epub 2018 Jul 24. Arch Virol. 2018. PMID: 30039318 Free PMC article. Review.
Cited by
-
Tracking pathogen transmission at the human-wildlife interface: banded mongoose and Escherichia coli.Ecohealth. 2013 Jun;10(2):115-28. doi: 10.1007/s10393-013-0838-2. Epub 2013 Apr 24. Ecohealth. 2013. PMID: 23612855
-
Current emerging SARS-CoV-2 pandemic: Potential direct/indirect negative impacts of virus persistence and related therapeutic drugs on the aquatic compartments.Environ Res. 2020 Sep;188:109808. doi: 10.1016/j.envres.2020.109808. Epub 2020 Jun 10. Environ Res. 2020. PMID: 32544725 Free PMC article.
-
Incidence of human adenoviruses and Hepatitis A virus in the final effluent of selected wastewater treatment plants in Eastern Cape Province, South Africa.Virol J. 2015 Jun 24;12:98. doi: 10.1186/s12985-015-0327-z. Virol J. 2015. PMID: 26104284 Free PMC article.
-
Application of next-generation sequencing technologies in virology.J Gen Virol. 2012 Sep;93(Pt 9):1853-1868. doi: 10.1099/vir.0.043182-0. Epub 2012 May 30. J Gen Virol. 2012. PMID: 22647373 Free PMC article. Review.
-
Exploring nucleo-cytoplasmic large DNA viruses in Tara Oceans microbial metagenomes.ISME J. 2013 Sep;7(9):1678-95. doi: 10.1038/ismej.2013.59. Epub 2013 Apr 11. ISME J. 2013. PMID: 23575371 Free PMC article.
References
-
- Culley AI, Lang AS, Suttle CA. 2006. Metagenomic analysis of coastal RNA virus communities. Science 312:1795–1798 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
