

Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors
- PMID: 21868758
- PMCID: PMC3209585
- DOI: 10.1158/0008-5472.CAN-11-0532
Intratumoral de novo steroid synthesis activates androgen receptor in castration-resistant prostate cancer and is upregulated by treatment with CYP17A1 inhibitors
Abstract
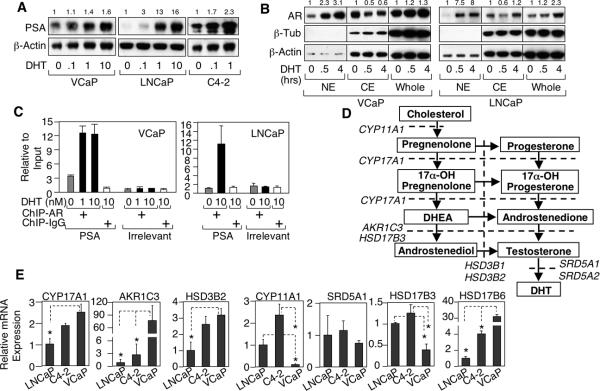
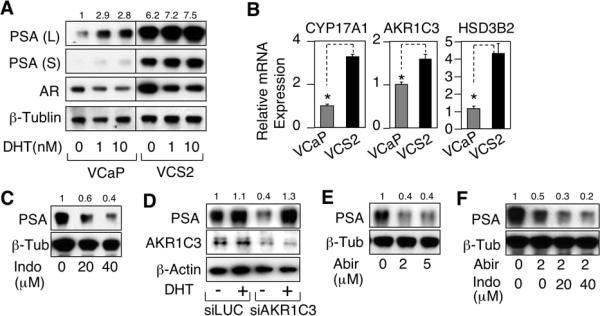
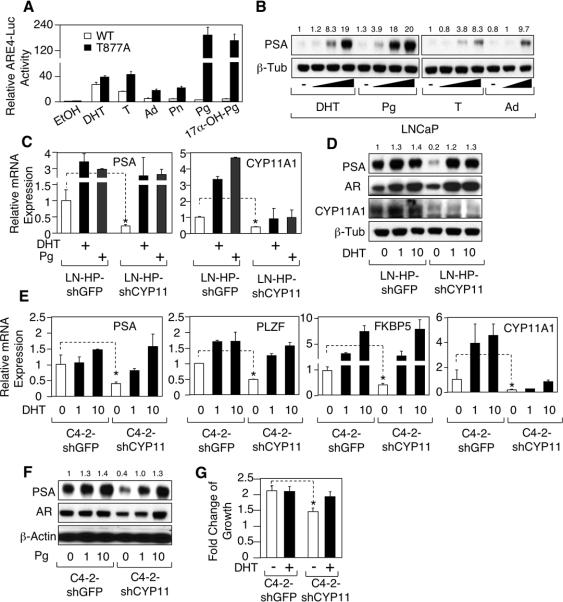
Relapse of castration-resistant prostate cancer (CRPC) that occurs after androgen deprivation therapy of primary prostate cancer can be mediated by reactivation of the androgen receptor (AR). One important mechanism mediating this AR reactivation is intratumoral conversion of the weak adrenal androgens DHEA and androstenedione into the AR ligands testosterone and dihydrotestosterone. DHEA and androstenedione are synthesized by the adrenals through the sequential actions of the cytochrome P450 enzymes CYP11A1 and CYP17A1, so that CYP17A1 inhibitors such as abiraterone are effective therapies for CRPC. However, the significance of intratumoral CYP17A1 and de novo androgen synthesis from cholesterol in CRPC, and the mechanisms contributing to CYP17A1 inhibitor resistance/relapse, remain to be determined. We report that AR activity in castration-resistant VCaP tumor xenografts can be restored through CYP17A1-dependent de novo androgen synthesis, and that abiraterone treatment of these xenografts imposes selective pressure for increased intratumoral expression of CYP17A1, thereby generating a mechanism for development of resistance to CYP17A1 inhibitors. Supporting the clinical relevance of this mechanism, we found that intratumoral expression of CYP17A1 was markedly increased in tumor biopsies from CRPC patients after CYP17A1 inhibitor therapy. We further show that CRPC cells expressing a progesterone responsive T877A mutant AR are not CYP17A1 dependent, but that AR activity in these cells is still steroid dependent and mediated by upstream CYP11A1-dependent intraturmoral pregnenolone/progesterone synthesis. Together, our results indicate that CRPCs resistant to CYP17A1 inhibition may remain steroid dependent and therefore responsive to therapies that can further suppress de novo intratumoral steroid synthesis.
Figures



Similar articles
-
A bypass mechanism of abiraterone-resistant prostate cancer: Accumulating CYP17A1 substrates activate androgen receptor signaling.Prostate. 2019 Jun;79(9):937-948. doi: 10.1002/pros.23799. Epub 2019 Apr 24. Prostate. 2019. PMID: 31017696 Free PMC article.
-
Resistance to CYP17A1 inhibition with abiraterone in castration-resistant prostate cancer: induction of steroidogenesis and androgen receptor splice variants.Clin Cancer Res. 2011 Sep 15;17(18):5913-25. doi: 10.1158/1078-0432.CCR-11-0728. Epub 2011 Aug 1. Clin Cancer Res. 2011. PMID: 21807635 Free PMC article.
-
Abiraterone switches castration-resistant prostate cancer dependency from adrenal androgens towards androgen receptor variants and glucocorticoid receptor signalling.Prostate. 2022 Apr;82(5):505-516. doi: 10.1002/pros.24297. Epub 2022 Jan 17. Prostate. 2022. PMID: 35037287 Free PMC article.
-
Intratumoral androgen biosynthesis in prostate cancer pathogenesis and response to therapy.Endocr Relat Cancer. 2011 Aug 30;18(5):R175-82. doi: 10.1530/ERC-10-0339. Print 2011 Oct. Endocr Relat Cancer. 2011. PMID: 21712345 Free PMC article. Review.
-
Androgen receptor functions in castration-resistant prostate cancer and mechanisms of resistance to new agents targeting the androgen axis.Oncogene. 2014 May 29;33(22):2815-25. doi: 10.1038/onc.2013.235. Epub 2013 Jun 10. Oncogene. 2014. PMID: 23752196 Free PMC article. Review.
Cited by
-
Androgen receptor signaling in circulating tumor cells as a marker of hormonally responsive prostate cancer.Cancer Discov. 2012 Nov;2(11):995-1003. doi: 10.1158/2159-8290.CD-12-0222. Epub 2012 Oct 23. Cancer Discov. 2012. PMID: 23093251 Free PMC article.
-
Targeting persistent androgen receptor signaling in castration-resistant prostate cancer.Med Oncol. 2016 May;33(5):44. doi: 10.1007/s12032-016-0759-3. Epub 2016 Apr 4. Med Oncol. 2016. PMID: 27042852 Review.
-
Niclosamide enhances abiraterone treatment via inhibition of androgen receptor variants in castration resistant prostate cancer.Oncotarget. 2016 May 31;7(22):32210-20. doi: 10.18632/oncotarget.8493. Oncotarget. 2016. PMID: 27049719 Free PMC article.
-
Interactions of abiraterone, eplerenone, and prednisolone with wild-type and mutant androgen receptor: a rationale for increasing abiraterone exposure or combining with MDV3100.Cancer Res. 2012 May 1;72(9):2176-82. doi: 10.1158/0008-5472.CAN-11-3980. Epub 2012 Mar 12. Cancer Res. 2012. PMID: 22411952 Free PMC article.
-
Human steroid biosynthesis for the oncologist.J Investig Med. 2012 Feb;60(2):495-503. doi: 10.2310/JIM.0b013e3182408567. J Investig Med. 2012. PMID: 22222232 Free PMC article. Review.
References
-
- Stanbrough M, Bubley GJ, Ross K, Golub TR, Rubin MA, Penning TM, et al. Increased expression of genes converting adrenal androgens to testosterone in androgen-independent prostate cancer. Cancer Res. 2006;66:2815–25. - PubMed
-
- Mendiratta P, Mostaghel E, Guinney J, Tewari AK, Porrello A, Barry WT, et al. Genomic strategy for targeting therapy in castration-resistant prostate cancer. J Clin Oncol. 2009;27:2022–9. - PubMed
-
- Scher HI, Sawyers CL. Biology of progressive, castration-resistant prostate cancer: directed therapies targeting the androgen-receptor signaling axis. J Clin Oncol. 2005;23:8253–61. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
