

Macrophage-elicited loss of estrogen receptor-α in breast cancer cells via involvement of MAPK and c-Jun at the ESR1 genomic locus
- PMID: 21860415
- PMCID: PMC3223561
- DOI: 10.1038/onc.2011.370
Macrophage-elicited loss of estrogen receptor-α in breast cancer cells via involvement of MAPK and c-Jun at the ESR1 genomic locus
Abstract
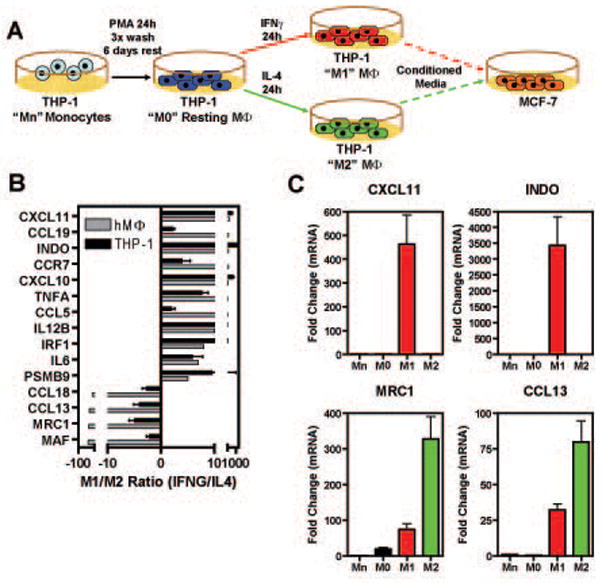
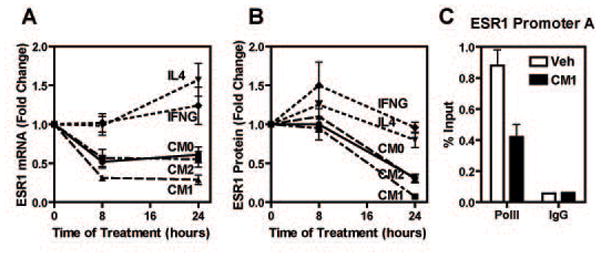
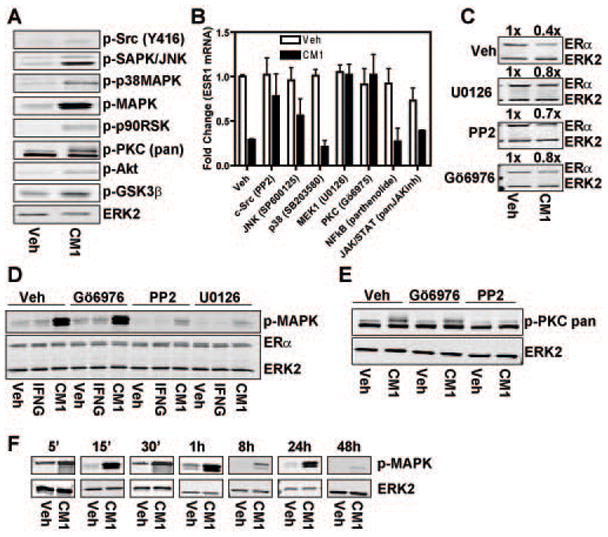
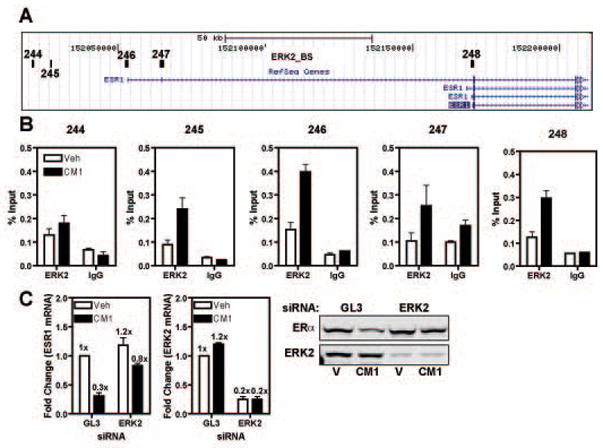
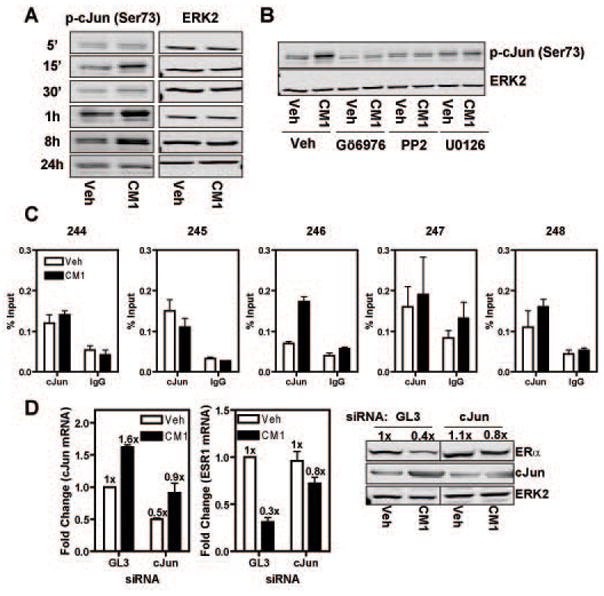
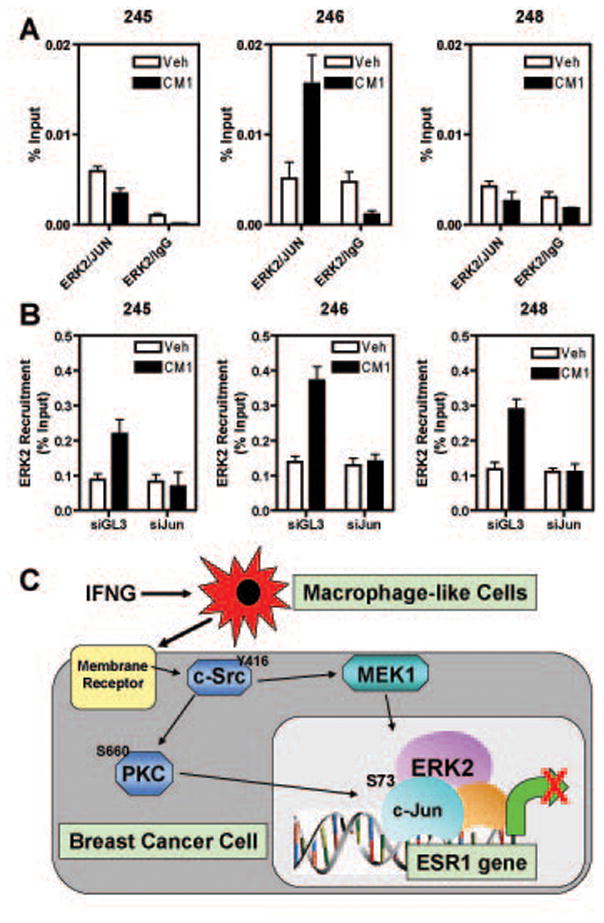
Estrogen receptor-α (ERα, ESR1) is a pivotal transcriptional regulator of breast cancer physiology and is targeted by endocrine therapies. Loss of ERα activity or expression is an indication of endocrine resistance and is associated with increased risk of tumor recurrence and worse prognosis. In this study, we sought to investigate whether elements of the tumor microenvironment, namely macrophages, would impact on ERα and we found that macrophage-derived factors caused loss of ERα expression in breast cancer cells. Conditioned media from macrophages caused activation of several intracellular pathways in breast cancer cells of which c-Src, protein kinase c and mitogen-activated protein kinase (MAPK) were essential for loss of ERα expression. Moreover, a prolonged hyperactivation of MAPK was observed. The activation of this kinase cascade resulted in recruitment of extracellular signal regulated kinase 2 (ERK2) directly to chromatin at the ESR1 gene locus in a process that was dependent upon activation and recruitment of the c-Jun transcription factor. Thus, we identify a novel mechanism for loss of ERα expression in breast cancer cells via macrophage activation of kinase cascades in the cancer cells causing transcriptional repression of the ESR1 gene by a direct chromatin action of a c-Jun/ERK2 complex. The findings in this study support an alternative mechanism, not intrinsic to the tumor cell but derived from the cross-talk with the tumor microenvironment, that could lead to endocrine resistance and might be targeted therapeutically to prevent loss of ERα expression in breast tumors.
Conflict of interest statement
F. S., Z. M. E, and B. S. K. have nothing to declare.
Figures
Similar articles
-
Activation of mitogen-activated protein kinase in estrogen receptor alpha-positive breast cancer cells in vitro induces an in vivo molecular phenotype of estrogen receptor alpha-negative human breast tumors.Cancer Res. 2006 Apr 1;66(7):3903-11. doi: 10.1158/0008-5472.CAN-05-4363. Cancer Res. 2006. PMID: 16585219
-
Hyperactivation of MAPK induces loss of ERalpha expression in breast cancer cells.Mol Endocrinol. 2001 Aug;15(8):1344-59. doi: 10.1210/mend.15.8.0678. Mol Endocrinol. 2001. PMID: 11463858
-
Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs.Mol Cell Biol. 2011 Jan;31(1):226-36. doi: 10.1128/MCB.00821-10. Epub 2010 Oct 18. Mol Cell Biol. 2011. PMID: 20956553 Free PMC article.
-
Reversal of the estrogen receptor negative phenotype in breast cancer and restoration of antiestrogen response.Clin Cancer Res. 2007 Dec 1;13(23):7029-36. doi: 10.1158/1078-0432.CCR-07-0587. Clin Cancer Res. 2007. PMID: 18056179
-
Estrogen Receptor Alpha Mutations, Truncations, Heterodimers, and Therapies.Endocrinology. 2024 Apr 29;165(6):bqae051. doi: 10.1210/endocr/bqae051. Endocrinology. 2024. PMID: 38643482 Review.
Cited by
-
Nur77 deficiency leads to systemic inflammation in elderly mice.J Inflamm (Lond). 2015 Jun 26;12:40. doi: 10.1186/s12950-015-0085-0. eCollection 2015. J Inflamm (Lond). 2015. PMID: 26113803 Free PMC article.
-
Targeting Obesity-Induced Macrophages during Preneoplastic Growth Promotes Mammary Epithelial Stem/Progenitor Activity, DNA Damage, and Tumor Formation.Cancer Res. 2020 Oct 15;80(20):4465-4475. doi: 10.1158/0008-5472.CAN-20-0789. Epub 2020 Aug 31. Cancer Res. 2020. PMID: 32868380 Free PMC article.
-
Estrogen receptor α promotes lung cancer cell invasion via increase of and cross-talk with infiltrated macrophages through the CCL2/CCR2/MMP9 and CXCL12/CXCR4 signaling pathways.Mol Oncol. 2020 Aug;14(8):1779-1799. doi: 10.1002/1878-0261.12701. Epub 2020 Jun 28. Mol Oncol. 2020. PMID: 32356397 Free PMC article.
-
Effect of macrophages on breast cancer cell proliferation, and on expression of hormone receptors, uPAR and HER-2.Int J Oncol. 2017 Jul;51(1):104-114. doi: 10.3892/ijo.2017.3996. Epub 2017 May 11. Int J Oncol. 2017. PMID: 28498427 Free PMC article.
-
Lung Tumor Cell-Derived Exosomes Promote M2 Macrophage Polarization.Cells. 2020 May 24;9(5):1303. doi: 10.3390/cells9051303. Cells. 2020. PMID: 32456301 Free PMC article.
References
-
- Allavena P, Sica A, Garlanda C, Mantovani A. Immunol Rev. 2008a;222:155–61. - PubMed
-
- Allavena P, Sica A, Solinas G, Porta C, Mantovani A. Crit Rev Oncol Hematol. 2008b;66:1–9. - PubMed
-
- Auwerx J. Experientia. 1991;47:22–31. - PubMed
-
- Bayliss J, Hilger A, Vishnu P, Diehl K, El-Ashry D. Clin Cancer Res. 2007;13:7029–36. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
