

Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome
- PMID: 21852578
- PMCID: PMC3169106
- DOI: 10.1073/pnas.1106015108
Proteasome assembly defect due to a proteasome subunit beta type 8 (PSMB8) mutation causes the autoinflammatory disorder, Nakajo-Nishimura syndrome
Abstract
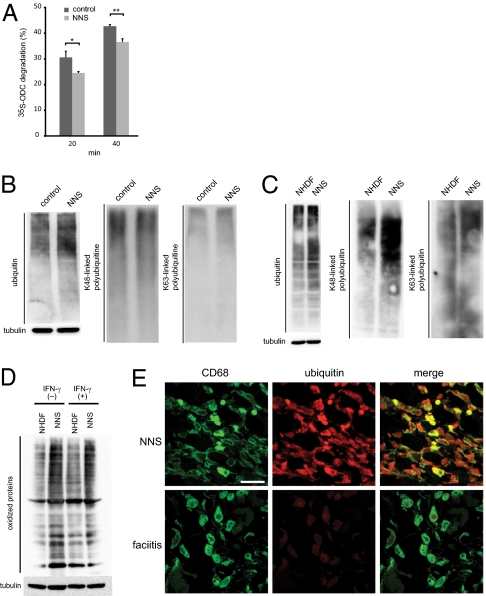
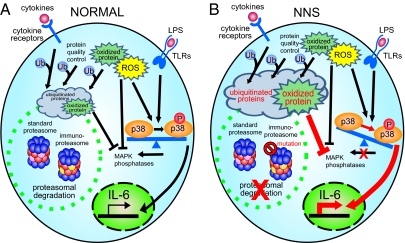
Nakajo-Nishimura syndrome (NNS) is a disorder that segregates in an autosomal recessive fashion. Symptoms include periodic fever, skin rash, partial lipomuscular atrophy, and joint contracture. Here, we report a mutation in the human proteasome subunit beta type 8 gene (PSMB8) that encodes the immunoproteasome subunit β5i in patients with NNS. This G201V mutation disrupts the β-sheet structure, protrudes from the loop that interfaces with the β4 subunit, and is in close proximity to the catalytic threonine residue. The β5i mutant is not efficiently incorporated during immunoproteasome biogenesis, resulting in reduced proteasome activity and accumulation of ubiquitinated and oxidized proteins within cells expressing immunoproteasomes. As a result, the level of interleukin (IL)-6 and IFN-γ inducible protein (IP)-10 in patient sera is markedly increased. Nuclear phosphorylated p38 and the secretion of IL-6 are increased in patient cells both in vitro and in vivo, which may account for the inflammatory response and periodic fever observed in these patients. These results show that a mutation within a proteasome subunit is the direct cause of a human disease and suggest that decreased proteasome activity can cause inflammation.
Conflict of interest statement
The authors declare no conflict of interest.
Figures



Similar articles
-
Nakajo-Nishimura syndrome and related proteasome-associated autoinflammatory syndromes.J Inflamm Res. 2019 Sep 17;12:259-265. doi: 10.2147/JIR.S194098. eCollection 2019. J Inflamm Res. 2019. PMID: 31576159 Free PMC article.
-
PSMB8 encoding the β5i proteasome subunit is mutated in joint contractures, muscle atrophy, microcytic anemia, and panniculitis-induced lipodystrophy syndrome.Am J Hum Genet. 2010 Dec 10;87(6):866-72. doi: 10.1016/j.ajhg.2010.10.031. Am J Hum Genet. 2010. PMID: 21129723 Free PMC article.
-
Nakajo-Nishimura syndrome: an autoinflammatory disorder showing pernio-like rashes and progressive partial lipodystrophy.Allergol Int. 2012 Jun;61(2):197-206. doi: 10.2332/allergolint.11-RAI-0416. Epub 2012 Mar 25. Allergol Int. 2012. PMID: 22441638 Review.
-
Myositis with sarcoplasmic inclusions in Nakajo-Nishimura syndrome: a genetic inflammatory myopathy.Neuropathol Appl Neurobiol. 2020 Oct;46(6):579-587. doi: 10.1111/nan.12614. Epub 2020 Mar 25. Neuropathol Appl Neurobiol. 2020. PMID: 32144790
-
[Nakajo-Nishimura syndrome].Nihon Rinsho Meneki Gakkai Kaishi. 2011;34(5):388-400. doi: 10.2177/jsci.34.388. Nihon Rinsho Meneki Gakkai Kaishi. 2011. PMID: 22041427 Review. Japanese.
Cited by
-
Role of Proteasomes in Inflammation.J Clin Med. 2021 Apr 20;10(8):1783. doi: 10.3390/jcm10081783. J Clin Med. 2021. PMID: 33923887 Free PMC article. Review.
-
Immunoproteasome deficiency modifies the alternative pathway of NFκB signaling.PLoS One. 2013;8(2):e56187. doi: 10.1371/journal.pone.0056187. Epub 2013 Feb 14. PLoS One. 2013. PMID: 23457524 Free PMC article.
-
Lighting the fires within: the cell biology of autoinflammatory diseases.Nat Rev Immunol. 2012 Jul 25;12(8):570-80. doi: 10.1038/nri3261. Nat Rev Immunol. 2012. PMID: 22828911 Free PMC article. Review.
-
Candidate Biomarkers and Molecular Mechanism Investigation for Glioblastoma Multiforme Utilizing WGCNA.Biomed Res Int. 2018 Sep 26;2018:4246703. doi: 10.1155/2018/4246703. eCollection 2018. Biomed Res Int. 2018. PMID: 30356407 Free PMC article.
-
Chronic atypical neutrophilic dermatosis with lipodystrophy and elevated temperature syndrome: a report of a novel mutation and review of the literature.Br J Dermatol. 2014 Jan;170(1):215-7. doi: 10.1111/bjd.12600. Br J Dermatol. 2014. PMID: 24001180 Free PMC article. Review. No abstract available.
References
-
- Nakajo A. Secondary hypertrophic osteoperiostosis with pernio. J Dermatol Urol. 1939;45:77–86.
-
- Nishimura N, Deki T, Kato S. Secondary hypertrophic osteoperiostosis with pernio-like skin lesions observed in two families. J Dermatol Venereol. 1950;60:136–141.
-
- Kitano Y, Matsunaga E, Morimoto T, Okada N, Sano S. A syndrome with nodular erythema, elongated and thickened fingers, and emaciation. Arch Dermatol. 1985;121:1053–1056. - PubMed
-
- Tanaka M, et al. Hereditary lipo-muscular atrophy with joint contracture, skin eruptions and hyper-gamma-globulinemia: A new syndrome. Intern Med. 1993;32:42–45. - PubMed
-
- Horikoshi A, Iwabuchi S, Iizuka Y, Hagiwara T, Amaki I. A case of partial lipodystrophy with erythema, dactylic deformities, calcification of the basal ganglia, immunological disorders, and low IQ level (Translated from Japanese) Rinsho Shinkeigaku. 1980;20:173–180. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
