

ATR autophosphorylation as a molecular switch for checkpoint activation
- PMID: 21777809
- PMCID: PMC3155885
- DOI: 10.1016/j.molcel.2011.06.019
ATR autophosphorylation as a molecular switch for checkpoint activation
Abstract
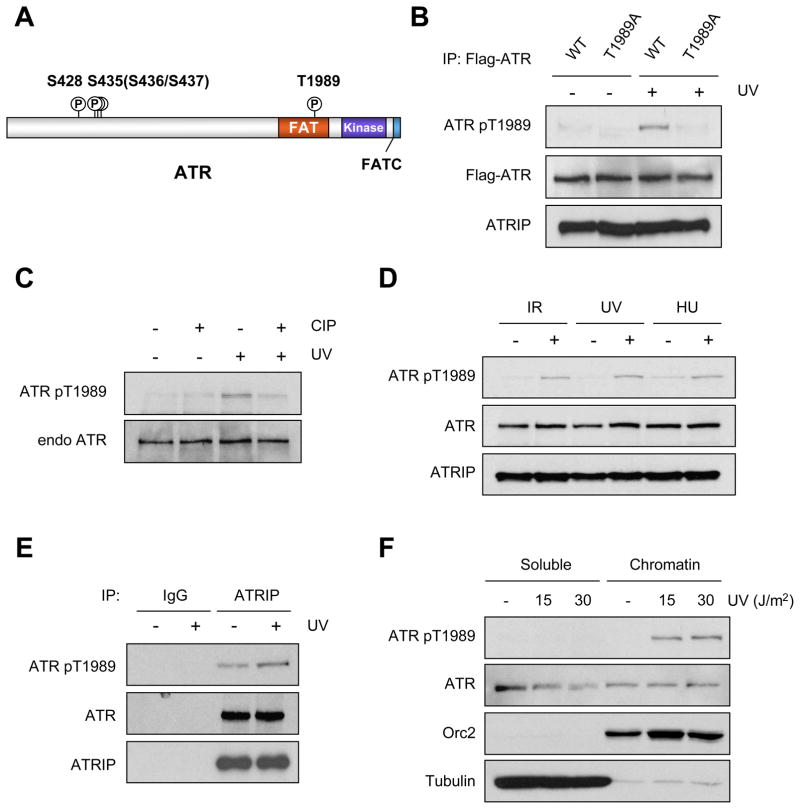
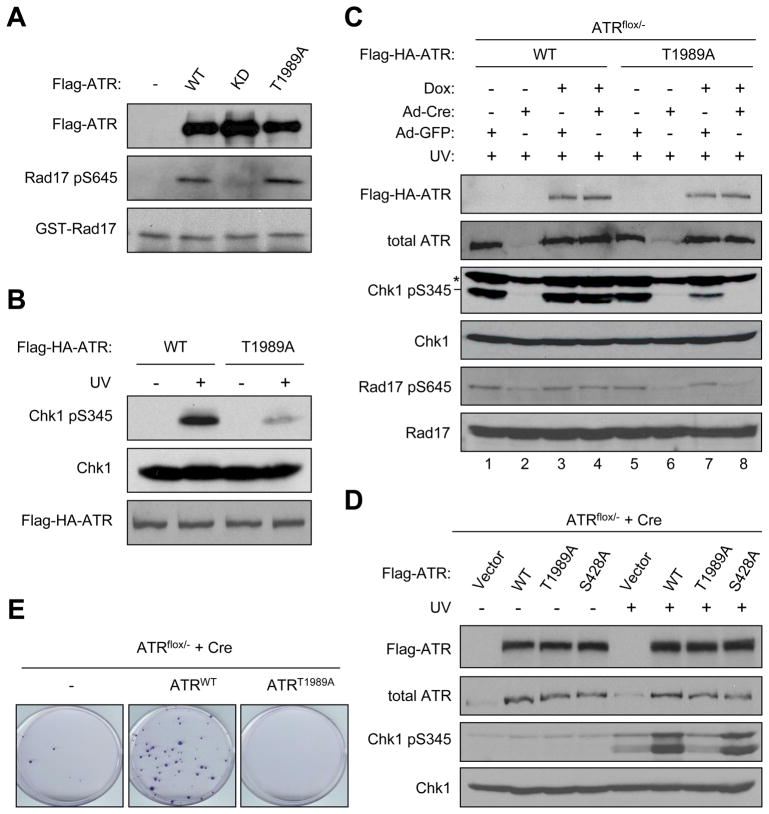
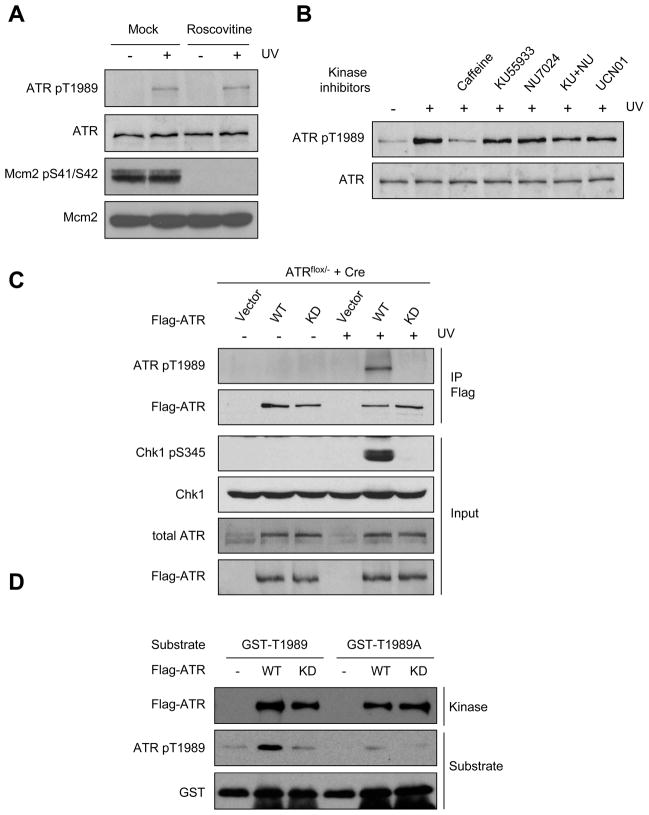
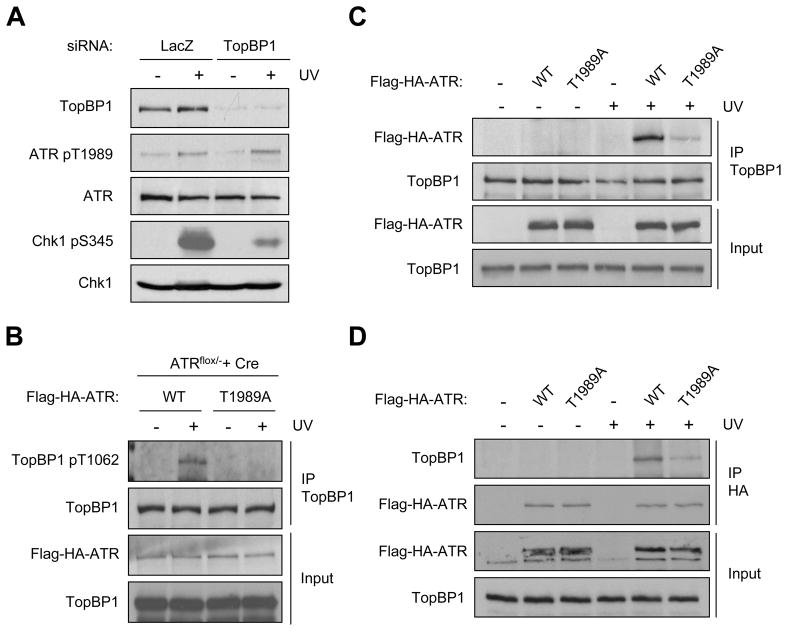
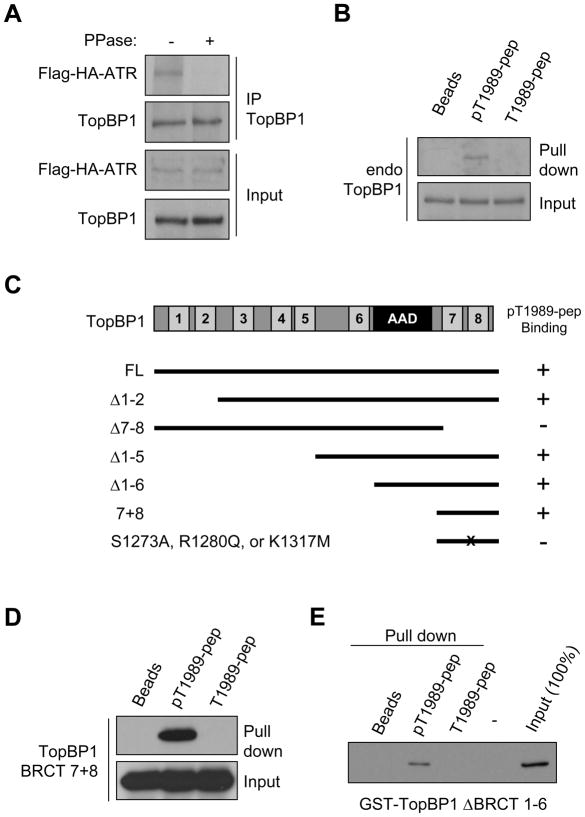
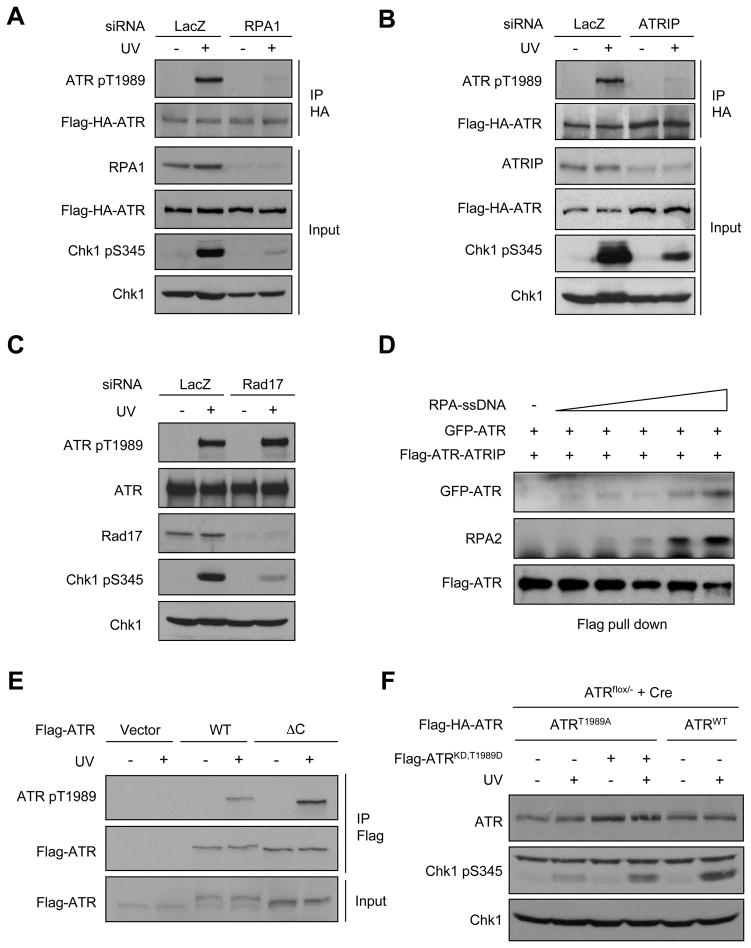
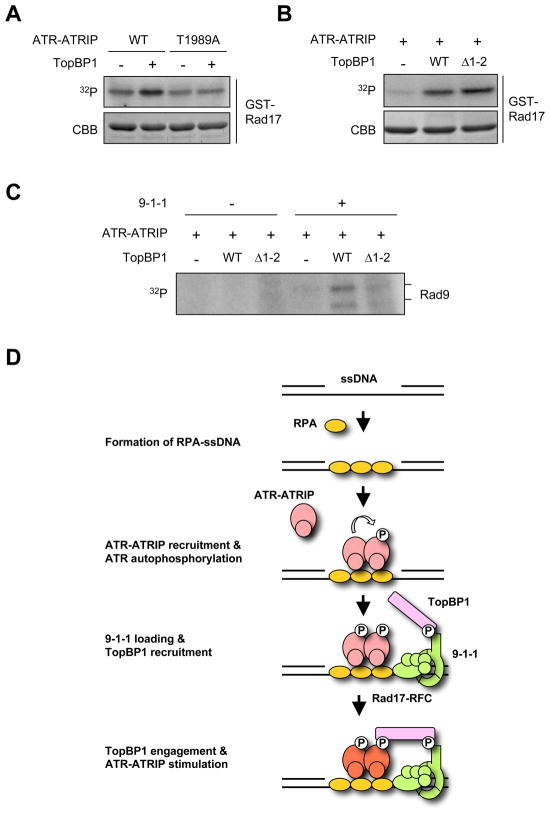
The ataxia telangiectasia-mutated and Rad3-related (ATR) kinase is a master checkpoint regulator safeguarding the genome. Upon DNA damage, the ATR-ATRIP complex is recruited to sites of DNA damage by RPA-coated single-stranded DNA and activated by an elusive process. Here, we show that ATR is transformed into a hyperphosphorylated state after DNA damage, and that a single autophosphorylation event at Thr 1989 is crucial for ATR activation. Phosphorylation of Thr 1989 relies on RPA, ATRIP, and ATR kinase activity, but unexpectedly not on the ATR stimulator TopBP1. Recruitment of ATR-ATRIP to RPA-ssDNA leads to congregation of ATR-ATRIP complexes and promotes Thr 1989 phosphorylation in trans. Phosphorylated Thr 1989 is directly recognized by TopBP1 via the BRCT domains 7 and 8, enabling TopBP1 to engage ATR-ATRIP, to stimulate the ATR kinase, and to facilitate ATR substrate recognition. Thus, ATR autophosphorylation on RPA-ssDNA is a molecular switch to launch robust checkpoint response.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
Reconstitution of RPA-covered single-stranded DNA-activated ATR-Chk1 signaling.Proc Natl Acad Sci U S A. 2010 Aug 3;107(31):13660-5. doi: 10.1073/pnas.1007856107. Epub 2010 Jun 28. Proc Natl Acad Sci U S A. 2010. PMID: 20616048 Free PMC article.
-
Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes.Science. 2003 Jun 6;300(5625):1542-8. doi: 10.1126/science.1083430. Science. 2003. PMID: 12791985
-
ATRIP Deacetylation by SIRT2 Drives ATR Checkpoint Activation by Promoting Binding to RPA-ssDNA.Cell Rep. 2016 Feb 16;14(6):1435-1447. doi: 10.1016/j.celrep.2016.01.018. Epub 2016 Feb 4. Cell Rep. 2016. PMID: 26854234 Free PMC article.
-
Activation of ATR-related protein kinase upon DNA damage recognition.Curr Genet. 2020 Apr;66(2):327-333. doi: 10.1007/s00294-019-01039-w. Epub 2019 Oct 17. Curr Genet. 2020. PMID: 31624858 Free PMC article. Review.
-
TopBP1 and DNA polymerase alpha-mediated recruitment of the 9-1-1 complex to stalled replication forks: implications for a replication restart-based mechanism for ATR checkpoint activation.Cell Cycle. 2009 Sep 15;8(18):2877-84. doi: 10.4161/cc.8.18.9485. Epub 2009 Sep 9. Cell Cycle. 2009. PMID: 19652550 Review.
Cited by
-
Kinase-dead ATM protein causes genomic instability and early embryonic lethality in mice.J Cell Biol. 2012 Aug 6;198(3):305-13. doi: 10.1083/jcb.201204098. J Cell Biol. 2012. PMID: 22869596 Free PMC article.
-
hMSH5 Facilitates the Repair of Camptothecin-induced Double-strand Breaks through an Interaction with FANCJ.J Biol Chem. 2015 Jul 24;290(30):18545-58. doi: 10.1074/jbc.M115.642884. Epub 2015 Jun 8. J Biol Chem. 2015. PMID: 26055704 Free PMC article.
-
Critical role of SMG7 in activation of the ATR-CHK1 axis in response to genotoxic stress.Sci Rep. 2021 Apr 5;11(1):7502. doi: 10.1038/s41598-021-86957-x. Sci Rep. 2021. PMID: 33820915 Free PMC article.
-
Functional mapping of PHF6 complexes in chromatin remodeling, replication dynamics, and DNA repair.Blood. 2022 Jun 9;139(23):3418-3429. doi: 10.1182/blood.2021014103. Blood. 2022. PMID: 35338774 Free PMC article.
-
An essential function for the ATR-activation-domain (AAD) of TopBP1 in mouse development and cellular senescence.PLoS Genet. 2013;9(8):e1003702. doi: 10.1371/journal.pgen.1003702. Epub 2013 Aug 8. PLoS Genet. 2013. PMID: 23950734 Free PMC article.
References
-
- Bakkenist CJ, Kastan MB. DNA damage activates ATM through intermolecular autophosphorylation and dimer dissociation. Nature. 2003;421:499–506. - PubMed
-
- Berkovich E, Monnat RJ, Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol. 2007;9:683–690. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
