

Cleavage of serum response factor mediated by enteroviral protease 2A contributes to impaired cardiac function
- PMID: 21769134
- PMCID: PMC3271589
- DOI: 10.1038/cr.2011.114
Cleavage of serum response factor mediated by enteroviral protease 2A contributes to impaired cardiac function
Abstract
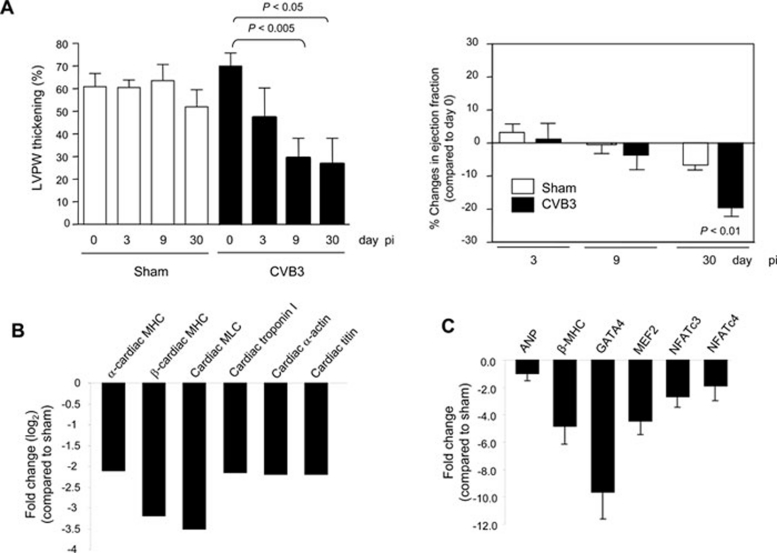
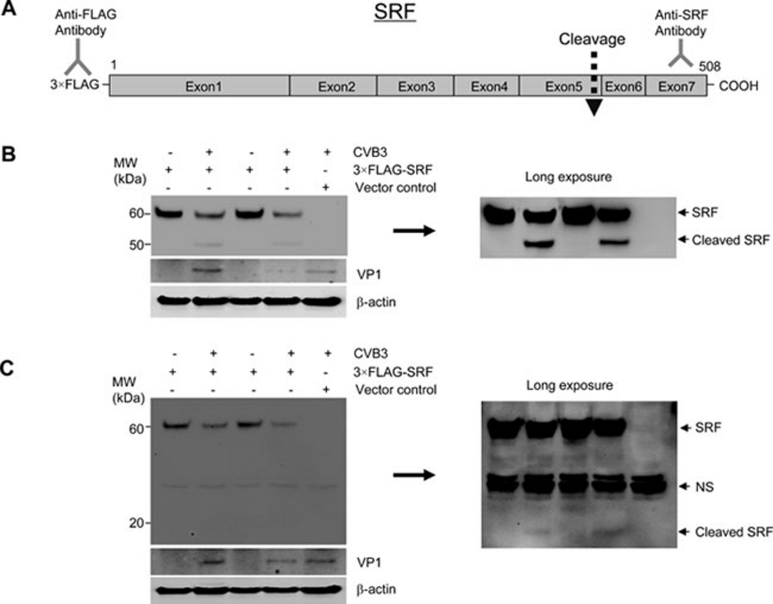
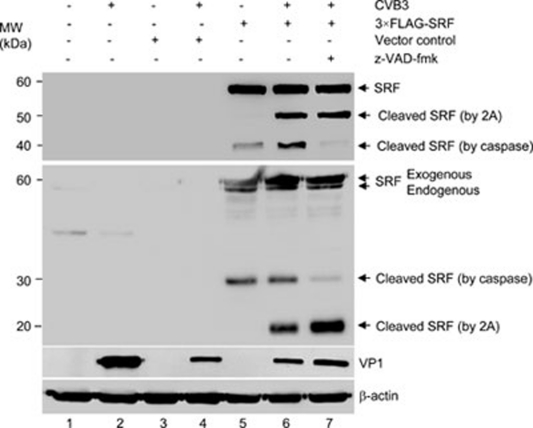
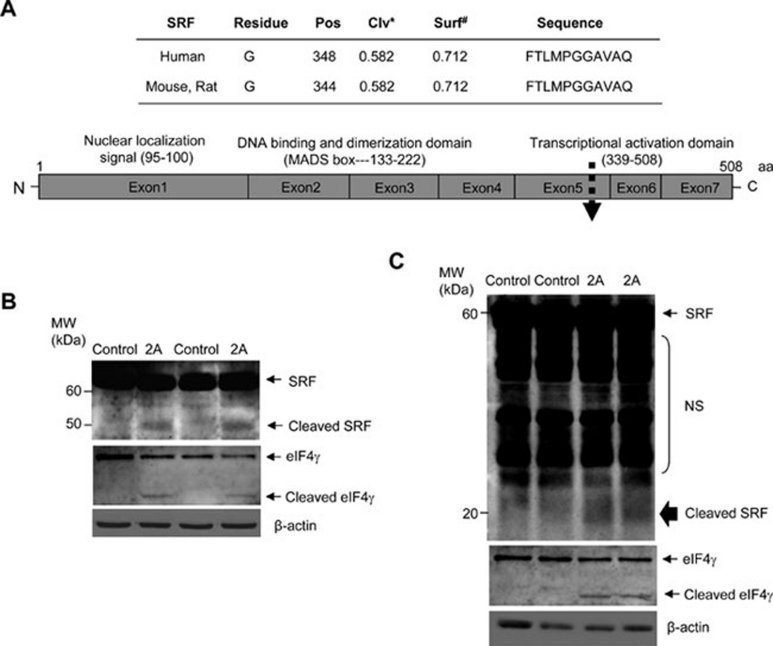
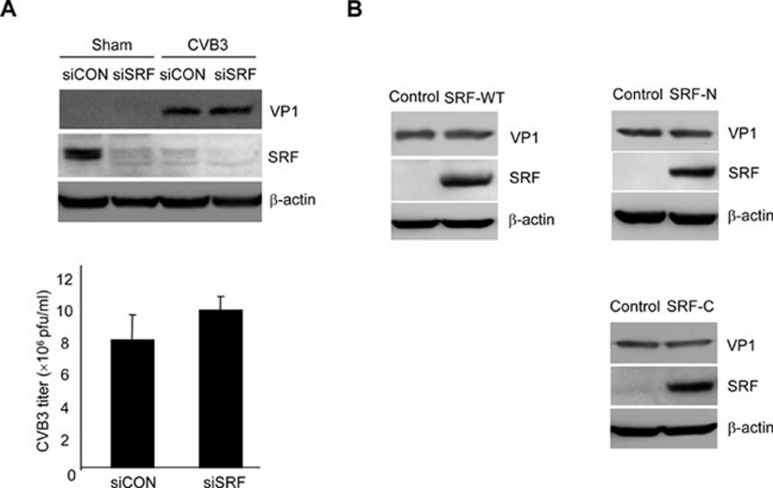
Enteroviral infection can lead to dilated cardiomyopathy (DCM), which is a major cause of cardiovascular mortality worldwide. However, the pathogenetic mechanisms have not been fully elucidated. Serum response factor (SRF) is a cardiac-enriched transcription regulator controlling the expression of a variety of target genes, including those involved in the contractile apparatus and immediate early response, as well as microRNAs that silence the expression of cardiac regulatory factors. Knockout of SRF in the heart results in downregulation of cardiac contractile gene expression and development of DCM. The goal of this study is to understand the role of SRF in enterovirus-induced cardiac dysfunction and progression to DCM. Here we report that SRF is cleaved following enteroviral infection of mouse heart and cultured cardiomyocytes. This cleavage is accompanied by impaired cardiac function and downregulation of cardiac-specific contractile and regulatory genes. Further investigation by antibody epitope mapping and site-directed mutagenesis demonstrates that SRF cleavage occurs at the region of its transactivation domain through the action of virus-encoded protease 2A. Moreover, we demonstrate that cleavage of SRF dissociates its transactivation domain from DNA-binding domain, resulting in the disruption of SRF-mediated gene transactivation. In addition to loss of functional SRF, finally we report that the N-terminal fragment of SRF cleavage products can also act as a dominant-negative transcription factor, which likely competes with the native SRF for DNA binding. Our results suggest a mechanism by which virus infection impairs heart function and may offer a new therapeutic strategy to ameliorate myocardial damage and progression to DCM.
Figures



Similar articles
-
Depressing time: Waiting, melancholia, and the psychoanalytic practice of care.In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. In: Kirtsoglou E, Simpson B, editors. The Time of Anthropology: Studies of Contemporary Chronopolitics. Abingdon: Routledge; 2020. Chapter 5. PMID: 36137063 Free Books & Documents. Review.
-
FUS/TLS Suppresses Enterovirus Replication and Promotes Antiviral Innate Immune Responses.J Virol. 2021 May 24;95(12):e00304-21. doi: 10.1128/JVI.00304-21. Print 2021 May 24. J Virol. 2021. PMID: 33827951 Free PMC article.
-
Enteroviral 3C protease cleaves N4BP1 to impair the host inflammatory response.J Virol. 2025 Jan 31;99(1):e0175824. doi: 10.1128/jvi.01758-24. Epub 2024 Dec 10. J Virol. 2025. PMID: 39655957 Free PMC article.
-
Far Posterior Approach for Rib Fracture Fixation: Surgical Technique and Tips.JBJS Essent Surg Tech. 2024 Dec 6;14(4):e23.00094. doi: 10.2106/JBJS.ST.23.00094. eCollection 2024 Oct-Dec. JBJS Essent Surg Tech. 2024. PMID: 39650795 Free PMC article.
-
Impact of residual disease as a prognostic factor for survival in women with advanced epithelial ovarian cancer after primary surgery.Cochrane Database Syst Rev. 2022 Sep 26;9(9):CD015048. doi: 10.1002/14651858.CD015048.pub2. Cochrane Database Syst Rev. 2022. PMID: 36161421 Free PMC article. Review.
Cited by
-
Cleavage of DAP5 by coxsackievirus B3 2A protease facilitates viral replication and enhances apoptosis by altering translation of IRES-containing genes.Cell Death Differ. 2016 May;23(5):828-40. doi: 10.1038/cdd.2015.145. Epub 2015 Nov 20. Cell Death Differ. 2016. PMID: 26586572 Free PMC article.
-
Mitochondria Dysfunction at the Heart of Viral Myocarditis: Mechanistic Insights and Therapeutic Implications.Viruses. 2023 Jan 26;15(2):351. doi: 10.3390/v15020351. Viruses. 2023. PMID: 36851568 Free PMC article. Review.
-
Approaches to identify and characterize microProteins and their potential uses in biotechnology.Cell Mol Life Sci. 2018 Jul;75(14):2529-2536. doi: 10.1007/s00018-018-2818-8. Epub 2018 Apr 18. Cell Mol Life Sci. 2018. PMID: 29670998 Free PMC article. Review.
-
Structures and Corresponding Functions of Five Types of Picornaviral 2A Proteins.Front Microbiol. 2017 Jul 21;8:1373. doi: 10.3389/fmicb.2017.01373. eCollection 2017. Front Microbiol. 2017. PMID: 28785248 Free PMC article. Review.
-
Phosphorylation and degradation of αB-crystallin during enterovirus infection facilitates viral replication and induces viral pathogenesis.Oncotarget. 2017 Aug 19;8(43):74767-74780. doi: 10.18632/oncotarget.20366. eCollection 2017 Sep 26. Oncotarget. 2017. PMID: 29088822 Free PMC article.
References
-
- Yajima T, Knowlton KU. Viral myocarditis: from the perspective of the virus. Circulation. 2009;119:2615–2624. - PubMed
-
- Xiong D, Yajima T, Lim BK, et al. Inducible cardiac-restricted expression of enteroviral protease 2A is sufficient to induce dilated cardiomyopathy. Circulation. 2007;115:94–102. - PubMed
-
- Badorff C, Lee GH, Lamphear BJ, et al. Enteroviral protease 2A cleaves dystrophin: evidence of cytoskeletal disruption in an acquired cardiomyopathy. Nat Med. 1999;5:320–326. - PubMed
-
- Deconinck AE, Rafael JA, Skinner JA, et al. Utrophin-dystrophin-deficient mice as a model for Duchenne muscular dystrophy. Cell. 1997;90:717–727. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
