

MyD88 deficiency ameliorates β-amyloidosis in an animal model of Alzheimer's disease
- PMID: 21763676
- PMCID: PMC3157279
- DOI: 10.1016/j.ajpath.2011.05.045
MyD88 deficiency ameliorates β-amyloidosis in an animal model of Alzheimer's disease
Abstract
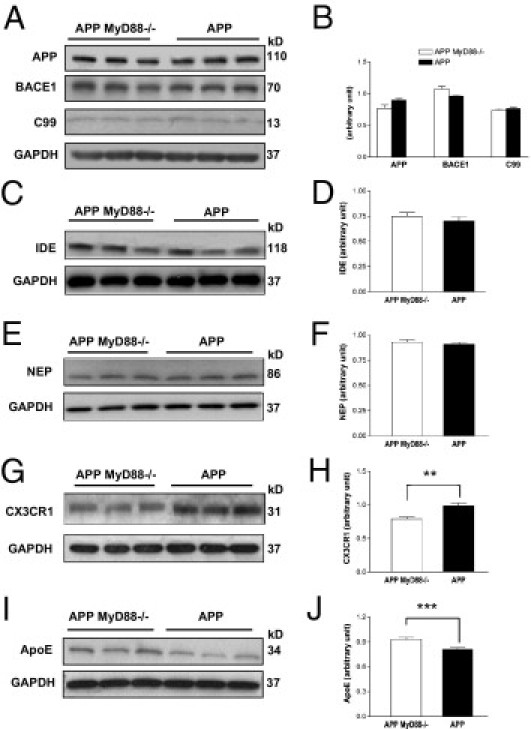
The accumulation of β-amyloid protein (Aβ) in the brain is thought to be a primary etiologic event in Alzheimer's disease (AD). Fibrillar Aβ plaques, a hallmark of AD abnormality, are closely associated with activated microglia. Activated microglia have contradictory roles in the pathogenesis of AD, being either neuroprotective (by clearing harmful Aβ and repairing damaged tissues) or neurotoxic (by producing proinflammatory cytokines and reactive oxygen species). Aβ aggregates can activate microglia by interacting with multiple toll-like receptors (TLRs), the pattern-recognition receptors of the innate immune system. Because the adapter protein MyD88 is essential for the downstream signaling of all TLRs, except TLR3, we investigated the effects of MyD88 deficiency (MyD88(-/-)) on Aβ accumulation and microglial activation in an AD mouse model. MyD88 deficiency decreased Aβ load and microglial activation in the brain. The decrease in Aβ load in an MyD88(-/-) AD mouse model was associated with increased and decreased protein expression of apolipoprotein E (apoE) and CX3CR1, respectively, compared with that in an MyD88 wild-type AD mouse model. These results suggest that MyD88 deficiency may reduce Aβ load by enhancing the phagocytic capability of microglia through fractalkine (the ligand of CX3CR1) signaling and by promoting apoE-mediated clearance of Aβ from the brain. These findings also suggest that chronic inflammatory responses induced by Aβ accumulation via the MyD88-dependent signaling pathway exacerbate β-amyloidosis in AD.
Copyright © 2011 American Society for Investigative Pathology. Published by Elsevier Inc. All rights reserved.
Figures


Similar articles
-
MyD88 is dispensable for cerebral amyloidosis and neuroinflammation in APP/PS1 transgenic mice.Am J Pathol. 2014 Nov;184(11):2855-61. doi: 10.1016/j.ajpath.2014.07.004. Epub 2014 Aug 28. Am J Pathol. 2014. PMID: 25174876 Free PMC article.
-
Effects of CX3CR1 and Fractalkine Chemokines in Amyloid Beta Clearance and p-Tau Accumulation in Alzheimer's Disease (AD) Rodent Models: Is Fractalkine a Systemic Biomarker for AD?Curr Alzheimer Res. 2016;13(4):403-12. doi: 10.2174/1567205013666151116125714. Curr Alzheimer Res. 2016. PMID: 26567742 Review.
-
CX3CR1 deficiency alters microglial activation and reduces beta-amyloid deposition in two Alzheimer's disease mouse models.Am J Pathol. 2010 Nov;177(5):2549-62. doi: 10.2353/ajpath.2010.100265. Epub 2010 Sep 23. Am J Pathol. 2010. PMID: 20864679 Free PMC article.
-
TLR4 mutation reduces microglial activation, increases Aβ deposits and exacerbates cognitive deficits in a mouse model of Alzheimer's disease.J Neuroinflammation. 2011 Aug 9;8:92. doi: 10.1186/1742-2094-8-92. J Neuroinflammation. 2011. PMID: 21827663 Free PMC article.
-
Role of apoe/Abeta interactions in the pathogenesis of Alzheimer's disease and cerebral amyloid angiopathy.J Mol Neurosci. 2001 Oct;17(2):147-55. doi: 10.1385/JMN:17:2:147. J Mol Neurosci. 2001. PMID: 11816788 Review.
Cited by
-
The effects of MyD88 deficiency on exploratory activity, anxiety, motor coordination, and spatial learning in C57BL/6 and APPswe/PS1dE9 mice.Behav Brain Res. 2012 Feb 1;227(1):36-42. doi: 10.1016/j.bbr.2011.10.027. Epub 2011 Oct 21. Behav Brain Res. 2012. PMID: 22051943 Free PMC article.
-
Eicosanoid receptor subtype-mediated opposing regulation of TLR-stimulated expression of astrocyte glial-derived neurotrophic factor.FASEB J. 2012 Jul;26(7):3075-83. doi: 10.1096/fj.11-200279. Epub 2012 Apr 12. FASEB J. 2012. PMID: 22499581 Free PMC article.
-
Fractalkine Mediates Communication between Pathogenic Proteins and Microglia: Implications of Anti-Inflammatory Treatments in Different Stages of Neurodegenerative Diseases.Int J Alzheimers Dis. 2012;2012:345472. doi: 10.1155/2012/345472. Epub 2012 Aug 5. Int J Alzheimers Dis. 2012. PMID: 22919540 Free PMC article.
-
Microglia in Alzheimer's Disease: A Role for Ion Channels.Front Neurosci. 2018 Sep 28;12:676. doi: 10.3389/fnins.2018.00676. eCollection 2018. Front Neurosci. 2018. PMID: 30323735 Free PMC article. Review.
-
Bioenergetic dysfunction and inflammation in Alzheimer's disease: a possible connection.Front Aging Neurosci. 2014 Nov 10;6:311. doi: 10.3389/fnagi.2014.00311. eCollection 2014. Front Aging Neurosci. 2014. PMID: 25426068 Free PMC article. Review.
References
-
- Hardy J., Selkoe D.J. The amyloid hypothesis of Alzheimer's disease: progress and problems on the road to therapeutics. Science. 2002;297:353–356. - PubMed
-
- Wyss-Coray T. Inflammation in Alzheimer disease: driving force, bystander or beneficial response? Nat Med. 2006;12:1005–1015. - PubMed
-
- Rivest S. Regulation of innate immune responses in the brain. Nat Rev Immunol. 2009;9:429–439. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
