

Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase
- PMID: 21705319
- PMCID: PMC3190678
- DOI: 10.1074/jbc.M111.248914
Thr-1989 phosphorylation is a marker of active ataxia telangiectasia-mutated and Rad3-related (ATR) kinase
Abstract
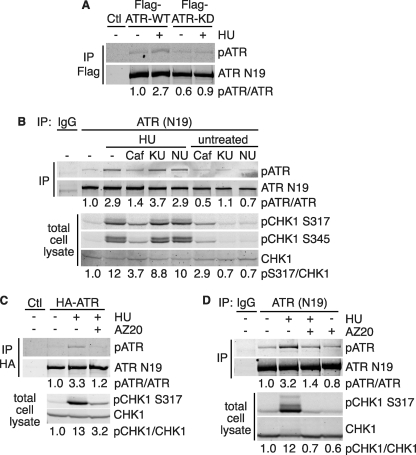
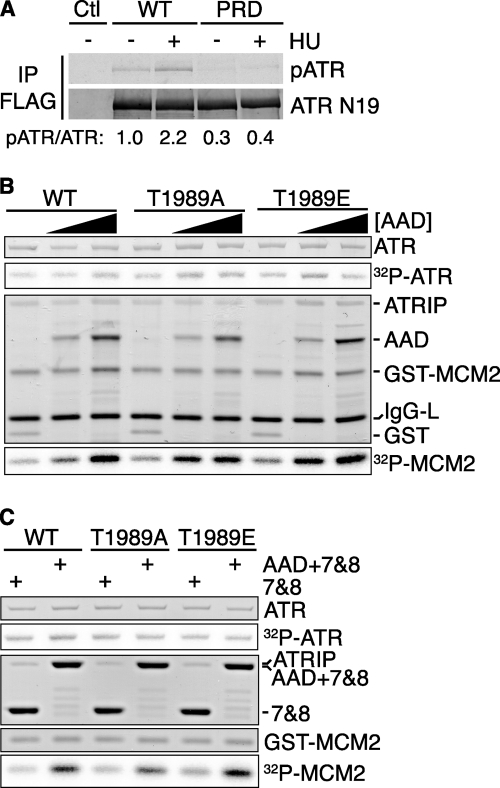
The DNA damage response kinases ataxia telangiectasia-mutated (ATM), DNA-dependent protein kinase (DNA-PK), and ataxia telangiectasia-mutated and Rad3-related (ATR) signal through multiple pathways to promote genome maintenance. These related kinases share similar methods of regulation, including recruitment to specific nucleic acid structures and association with protein activators. ATM and DNA-PK also are regulated via phosphorylation, which provides a convenient biomarker for their activity. Whether phosphorylation regulates ATR is unknown. Here we identify ATR Thr-1989 as a DNA damage-regulated phosphorylation site. Selective inhibition of ATR prevents Thr-1989 phosphorylation, and phosphorylation requires ATR activation. Cells engineered to express only a non-phosphorylatable T1989A mutant exhibit a modest ATR functional defect. Our results suggest that, like ATM and DNA-PK, phosphorylation regulates ATR, and phospho-peptide specific antibodies to Thr-1989 provide a proximal marker of ATR activation.
Figures




Similar articles
-
Tethering DNA damage checkpoint mediator proteins topoisomerase IIbeta-binding protein 1 (TopBP1) and Claspin to DNA activates ataxia-telangiectasia mutated and RAD3-related (ATR) phosphorylation of checkpoint kinase 1 (Chk1).J Biol Chem. 2011 Jun 3;286(22):19229-36. doi: 10.1074/jbc.M111.237958. Epub 2011 Apr 18. J Biol Chem. 2011. PMID: 21502314 Free PMC article.
-
Ataxia-telangiectasia mutated (ATM)-dependent activation of ATR occurs through phosphorylation of TopBP1 by ATM.J Biol Chem. 2007 Jun 15;282(24):17501-6. doi: 10.1074/jbc.M701770200. Epub 2007 Apr 19. J Biol Chem. 2007. PMID: 17446169
-
Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage.J Biol Chem. 2005 Jan 14;280(2):1186-92. doi: 10.1074/jbc.M410873200. Epub 2004 Nov 8. J Biol Chem. 2005. PMID: 15533933
-
The role of NBS1 in the modulation of PIKK family proteins ATM and ATR in the cellular response to DNA damage.Cancer Lett. 2006 Nov 8;243(1):9-15. doi: 10.1016/j.canlet.2006.01.026. Epub 2006 Mar 10. Cancer Lett. 2006. PMID: 16530324 Free PMC article. Review.
-
Structure and function of the apical PIKKs in double-strand break repair.Curr Opin Struct Biol. 2023 Oct;82:102651. doi: 10.1016/j.sbi.2023.102651. Epub 2023 Jul 10. Curr Opin Struct Biol. 2023. PMID: 37437397 Free PMC article. Review.
Cited by
-
ATR prevents Ca2+ overload-induced necrotic cell death through phosphorylation-mediated inactivation of PARP1 without DNA damage signaling.FASEB J. 2021 May;35(5):e21373. doi: 10.1096/fj.202001636RRR. FASEB J. 2021. PMID: 33811702 Free PMC article.
-
Mec1ATR Autophosphorylation and Ddc2ATRIP Phosphorylation Regulates DNA Damage Checkpoint Signaling.Cell Rep. 2019 Jul 23;28(4):1090-1102.e3. doi: 10.1016/j.celrep.2019.06.068. Cell Rep. 2019. PMID: 31340146 Free PMC article.
-
Inhibiting Wee1 and ATR kinases produces tumor-selective synthetic lethality and suppresses metastasis.J Clin Invest. 2019 Mar 1;129(3):1329-1344. doi: 10.1172/JCI122622. Epub 2019 Feb 18. J Clin Invest. 2019. PMID: 30645202 Free PMC article.
-
Radiation-dose-dependent functional synergisms between ATM, ATR and DNA-PKcs in checkpoint control and resection in G2-phase.Sci Rep. 2019 Jun 4;9(1):8255. doi: 10.1038/s41598-019-44771-6. Sci Rep. 2019. PMID: 31164689 Free PMC article.
-
RPA inhibition increases replication stress and suppresses tumor growth.Cancer Res. 2014 Sep 15;74(18):5165-72. doi: 10.1158/0008-5472.CAN-14-0306. Epub 2014 Jul 28. Cancer Res. 2014. PMID: 25070753 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
