

Galectin-9 binding to cell surface protein disulfide isomerase regulates the redox environment to enhance T-cell migration and HIV entry
- PMID: 21670307
- PMCID: PMC3127870
- DOI: 10.1073/pnas.1017954108
Galectin-9 binding to cell surface protein disulfide isomerase regulates the redox environment to enhance T-cell migration and HIV entry
Abstract
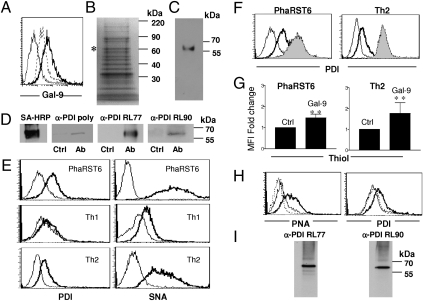
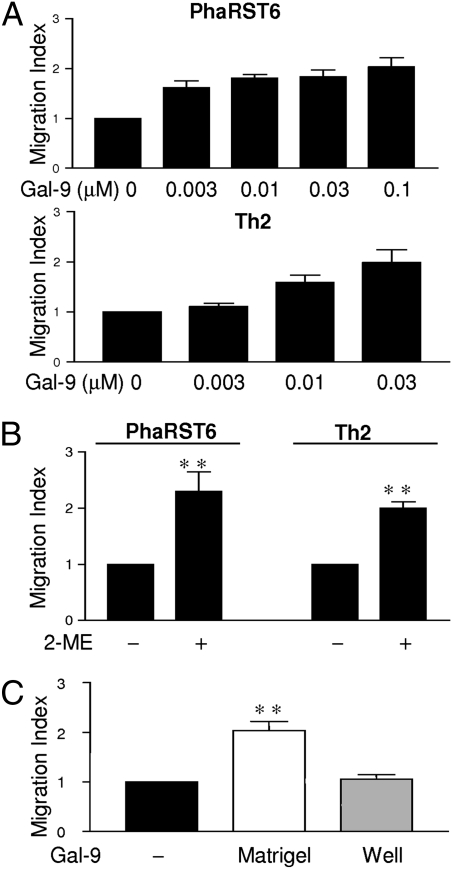
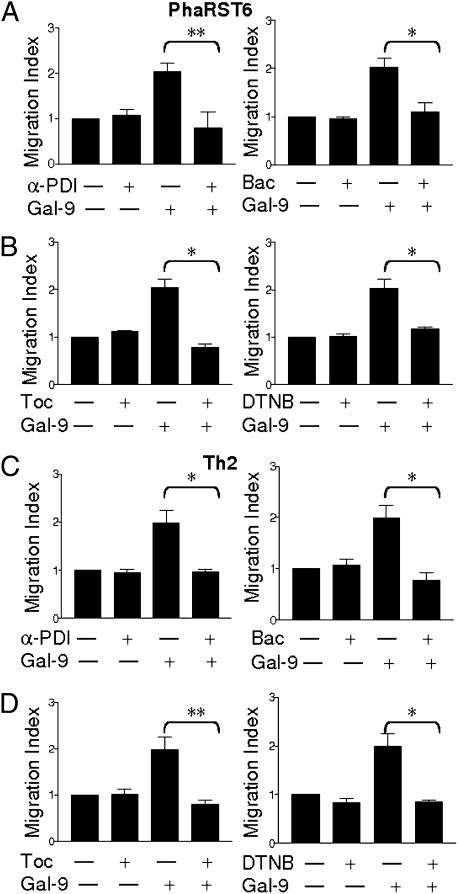
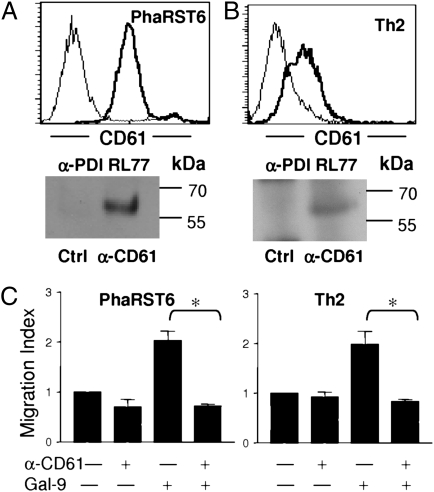
Interaction of cell surface glycoproteins with endogenous lectins on the cell surface regulates formation and maintenance of plasma membrane domains, clusters signaling complexes, and controls the residency time of glycoproteins on the plasma membrane. Galectin-9 is a soluble, secreted lectin that binds to glycoprotein receptors to form galectin-glycoprotein lattices on the cell surface. Whereas galectin-9 binding to specific glycoprotein receptors induces death of CD4 Th1 cells, CD4 Th2 cells are resistant to galectin-9 death due to alternative glycosylation. On Th2 cells, galectin-9 binds cell surface protein disulfide isomerase (PDI), increasing retention of PDI on the cell surface and altering the redox status at the plasma membrane. Cell surface PDI regulates integrin function on platelets and also enhances susceptibility of T cells to infection with HIV. We find that galectin-9 binding to PDI on Th2 cells results in increased cell migration through extracellular matrix via β3 integrins, identifying a unique mechanism to regulate T-cell migration. In addition, galectin-9 binding to PDI on T cells potentiates infection with HIV. We identify a mechanism for regulating cell surface redox status via a galectin-glycoprotein lattice, to regulate distinct T-cell functions.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

Similar articles
-
Galectin-9 binds to O-glycans on protein disulfide isomerase.Glycobiology. 2017 Sep 1;27(9):878-887. doi: 10.1093/glycob/cwx065. Glycobiology. 2017. PMID: 28810662 Free PMC article.
-
The catalytic activity of protein disulfide isomerase is involved in human immunodeficiency virus envelope-mediated membrane fusion after CD4 cell binding.J Infect Dis. 2001 Mar 1;183(5):744-52. doi: 10.1086/318823. Epub 2001 Jan 25. J Infect Dis. 2001. PMID: 11181151
-
Thiol/disulfide exchange is a prerequisite for CXCR4-tropic HIV-1 envelope-mediated T-cell fusion during viral entry.Blood. 2004 Mar 1;103(5):1586-94. doi: 10.1182/blood-2003-05-1390. Epub 2003 Oct 30. Blood. 2004. PMID: 14592831
-
Galectin-glycan lattices regulate cell-surface glycoprotein organization and signalling.Biochem Soc Trans. 2008 Dec;36(Pt 6):1472-7. doi: 10.1042/BST0361472. Biochem Soc Trans. 2008. PMID: 19021578 Free PMC article. Review.
-
T-cell growth, cell surface organization, and the galectin-glycoprotein lattice.Immunol Rev. 2009 Jul;230(1):232-46. doi: 10.1111/j.1600-065X.2009.00796.x. Immunol Rev. 2009. PMID: 19594640 Free PMC article. Review.
Cited by
-
Blockage of Galectin-receptor Interactions by α-lactose Exacerbates Plasmodium berghei-induced Pulmonary Immunopathology.Sci Rep. 2016 Aug 24;6:32024. doi: 10.1038/srep32024. Sci Rep. 2016. PMID: 27554340 Free PMC article.
-
Impact of Exogenous Galectin-9 on Human T Cells: CONTRIBUTION OF THE T CELL RECEPTOR COMPLEX TO ANTIGEN-INDEPENDENT ACTIVATION BUT NOT TO APOPTOSIS INDUCTION.J Biol Chem. 2015 Jul 3;290(27):16797-811. doi: 10.1074/jbc.M115.661272. Epub 2015 May 6. J Biol Chem. 2015. PMID: 25947381 Free PMC article.
-
Galectin-9 - ligand axis: an emerging therapeutic target for multiple myeloma.Front Immunol. 2024 Sep 25;15:1469794. doi: 10.3389/fimmu.2024.1469794. eCollection 2024. Front Immunol. 2024. PMID: 39386209 Free PMC article. Review.
-
Protein Oxidative Damage in UV-Related Skin Cancer and Dysplastic Lesions Contributes to Neoplastic Promotion and Progression.Cancers (Basel). 2020 Jan 1;12(1):110. doi: 10.3390/cancers12010110. Cancers (Basel). 2020. PMID: 31906275 Free PMC article.
-
Modulation of the Gal-9/TIM-3 Immune Checkpoint with α-Lactose. Does Anomery of Lactose Matter?Cancers (Basel). 2021 Dec 18;13(24):6365. doi: 10.3390/cancers13246365. Cancers (Basel). 2021. PMID: 34944985 Free PMC article. Review.
References
-
- Jordan PA, Gibbins JM. Extracellular disulfide exchange and the regulation of cellular function. Antioxid Redox Signal. 2006;8:312–324. - PubMed
-
- Hogg PJ. Disulfide bonds as switches for protein function. Trends Biochem Sci. 2003;28:210–214. - PubMed
-
- Lawrence DA, Song R, Weber P. Surface thiols of human lymphocytes and their changes after in vitro and in vivo activation. J Leukoc Biol. 1996;60:611–618. - PubMed
-
- Turano C, Coppari S, Altieri F, Ferraro A. Proteins of the PDI family: Unpredicted non-ER locations and functions. J Cell Physiol. 2002;193:154–163. - PubMed
-
- Fenouillet E, Barbouche R, Courageot J, Miquelis R. The catalytic activity of protein disulfide isomerase is involved in human immunodeficiency virus envelope-mediated membrane fusion after CD4 cell binding. J Infect Dis. 2001;183:744–752. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
