

MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness
- PMID: 21637773
- PMCID: PMC3102730
- DOI: 10.1371/journal.ppat.1002070
MDA5 and TLR3 initiate pro-inflammatory signaling pathways leading to rhinovirus-induced airways inflammation and hyperresponsiveness
Abstract
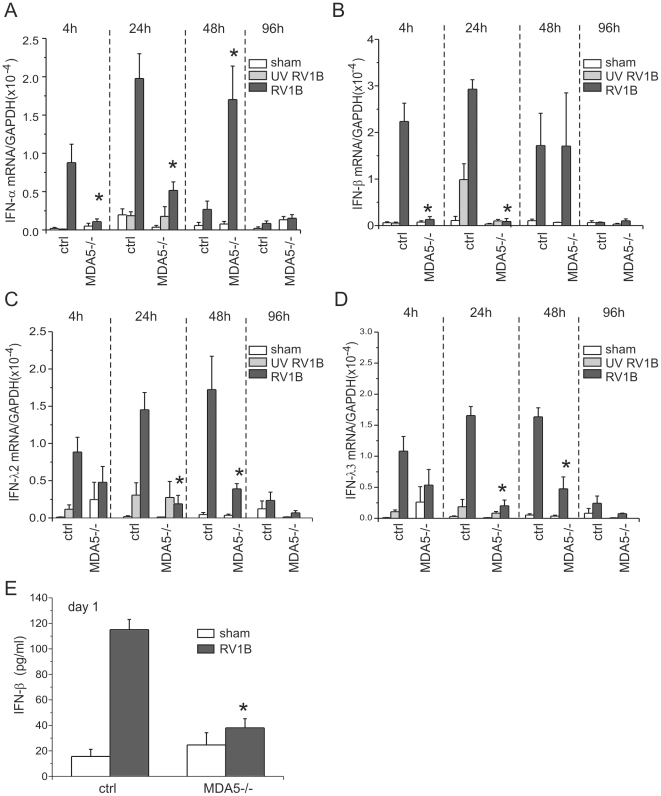
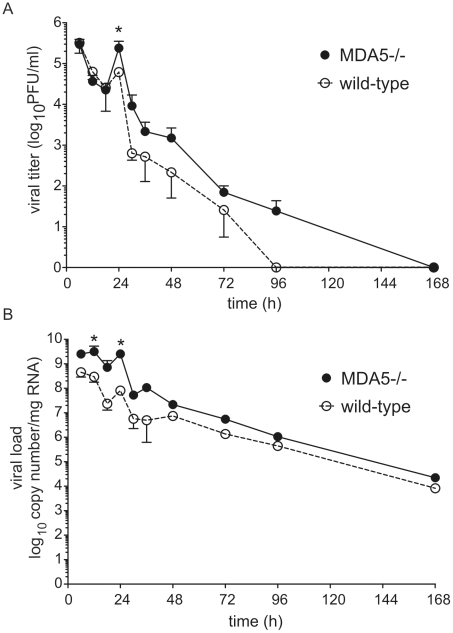
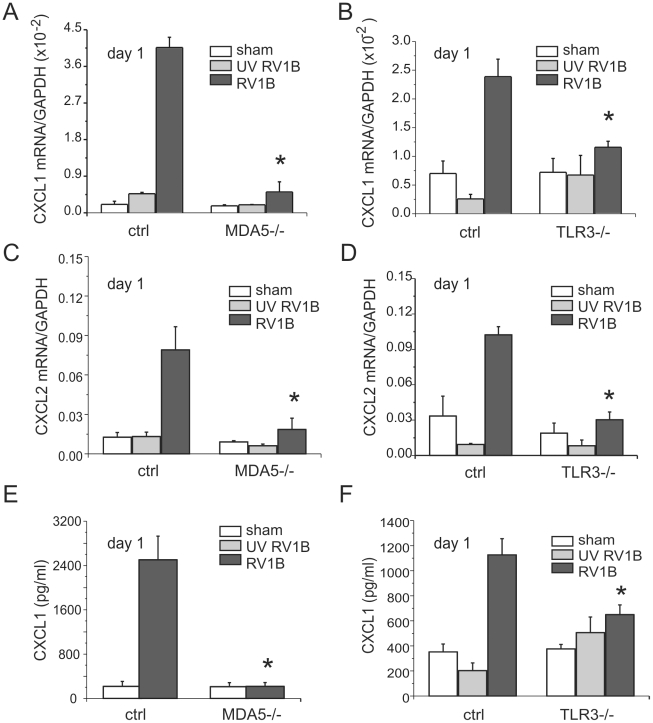
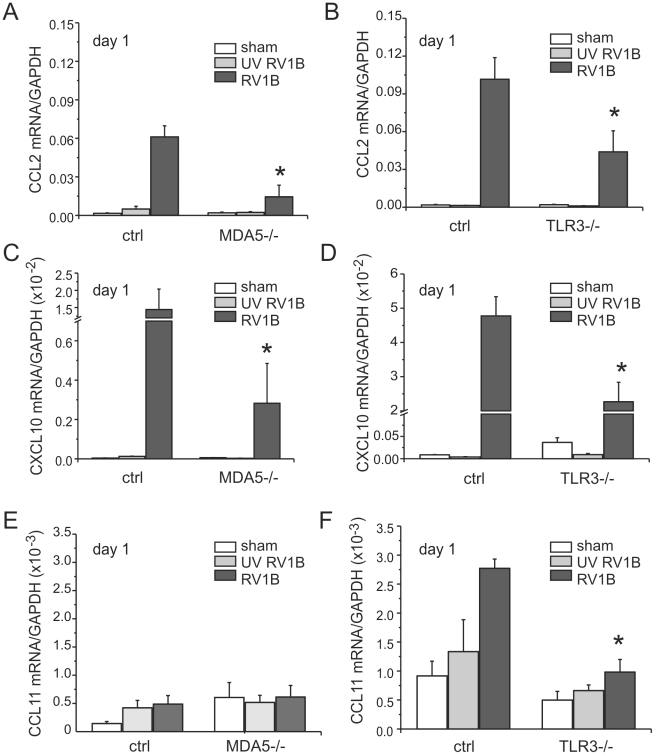
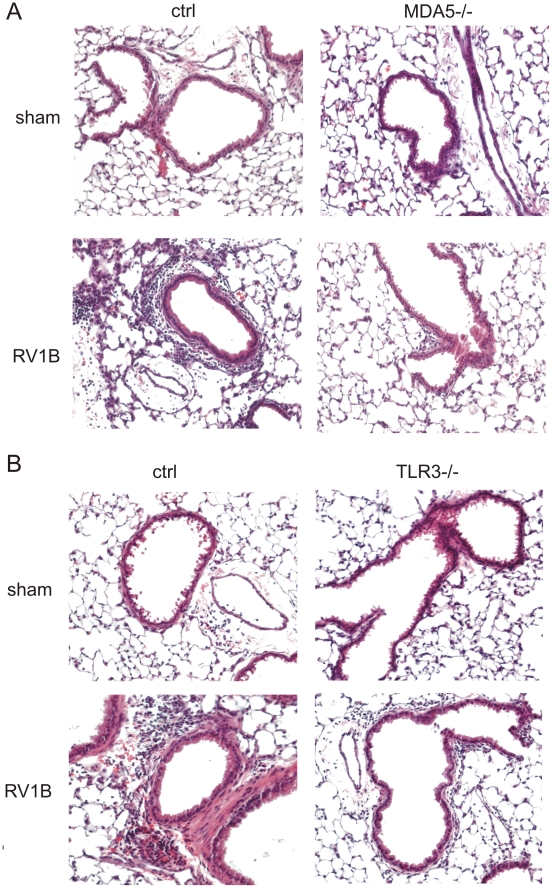
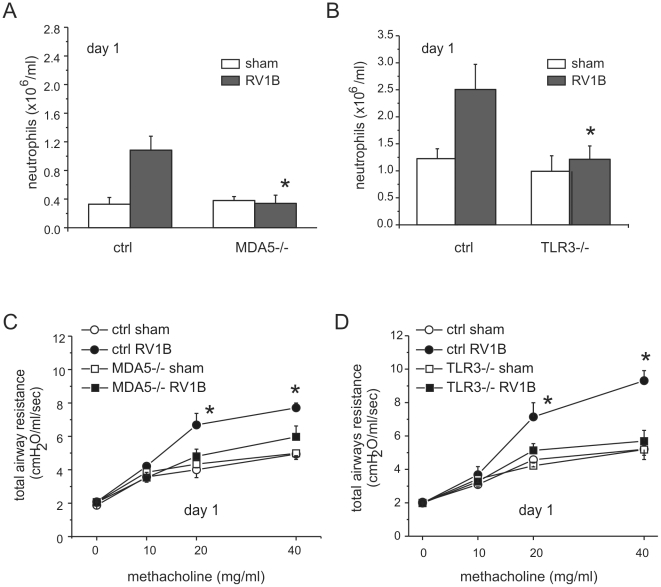
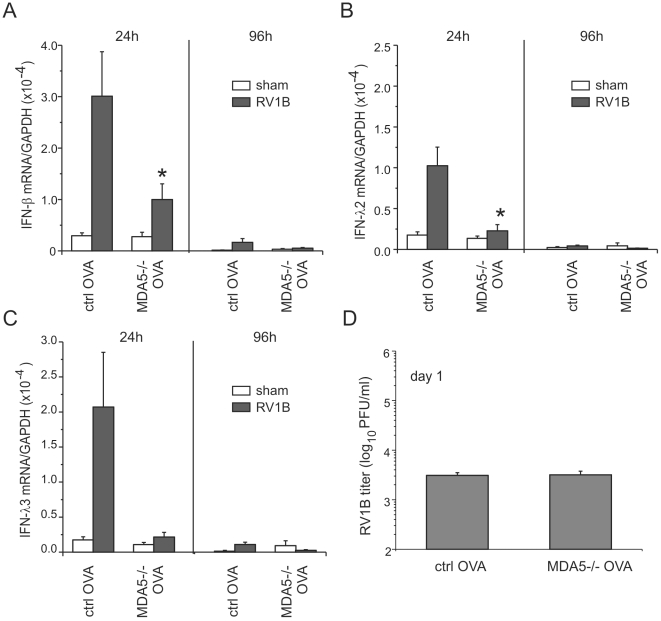
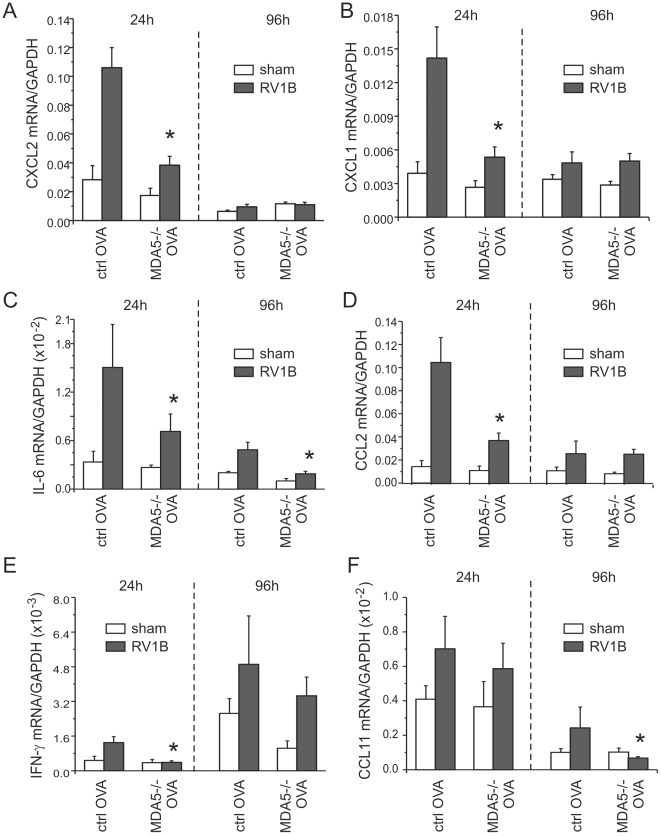
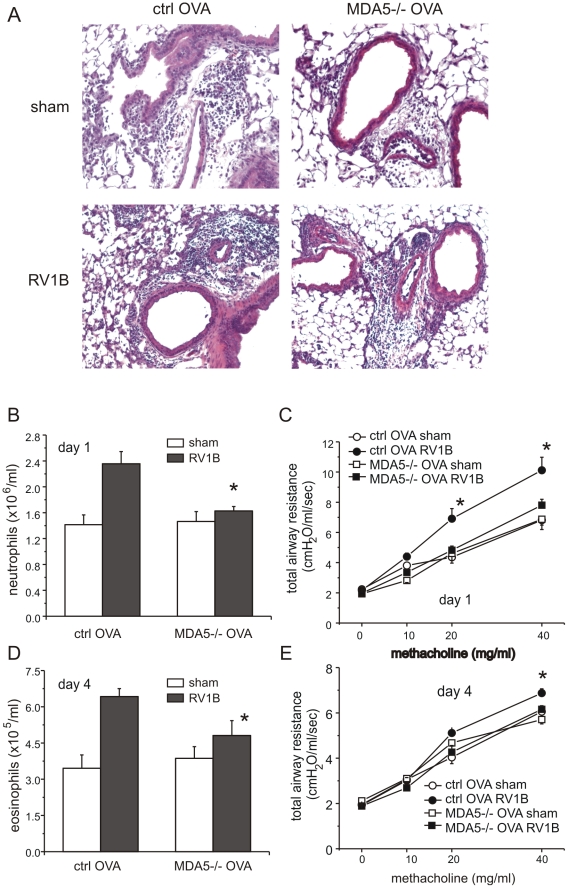
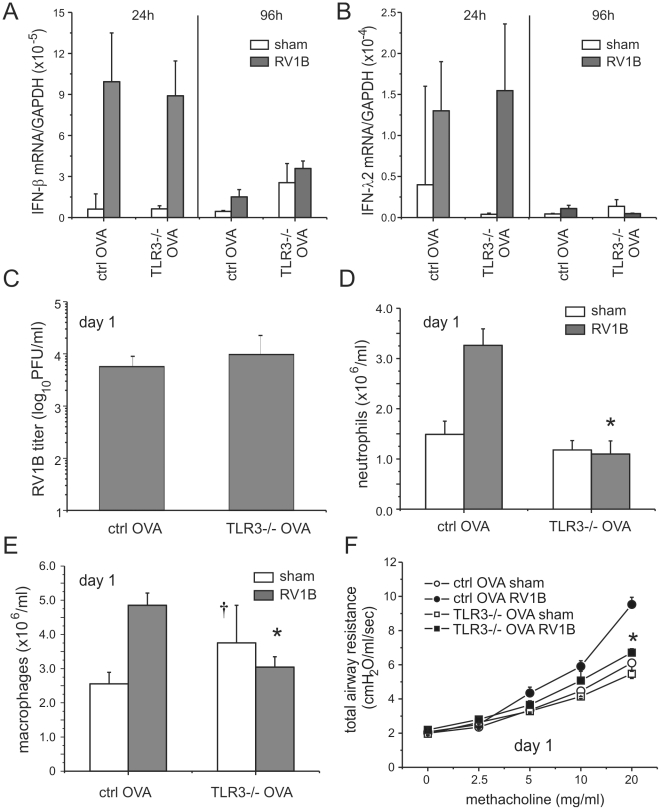
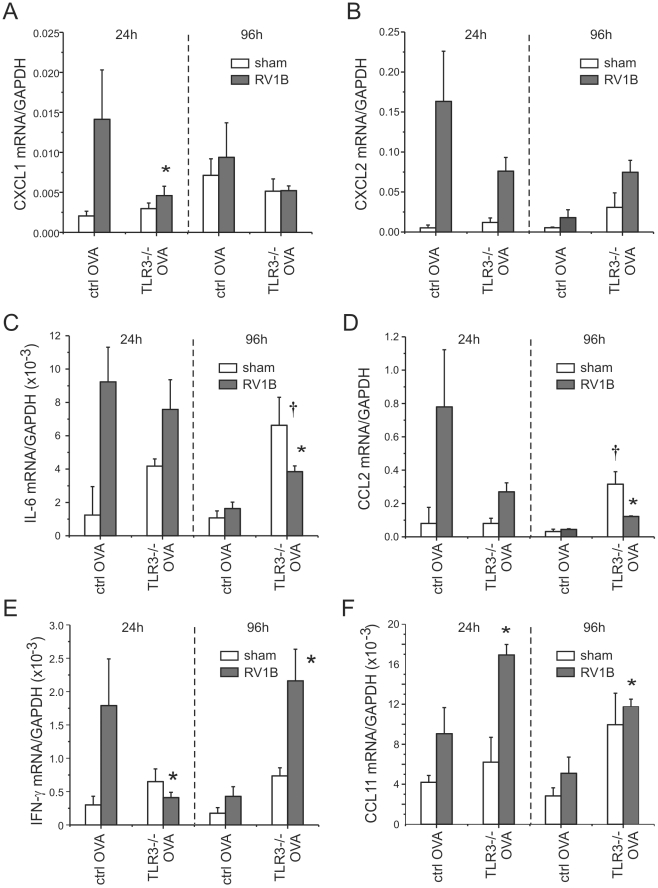
Rhinovirus (RV), a single-stranded RNA picornavirus, is the most frequent cause of asthma exacerbations. We previously demonstrated in human bronchial epithelial cells that melanoma differentiation-associated gene (MDA)-5 and the adaptor protein for Toll-like receptor (TLR)-3 are each required for maximal RV1B-induced interferon (IFN) responses. However, in vivo, the overall airway response to viral infection likely represents a coordinated response integrating both antiviral and pro-inflammatory pathways. We examined the airway responses of MDA5- and TLR3-deficient mice to infection with RV1B, a minor group virus which replicates in mouse lungs. MDA5 null mice showed a delayed type I IFN and attenuated type III IFN response to RV1B infection, leading to a transient increase in viral titer. TLR3 null mice showed normal IFN responses and unchanged viral titers. Further, RV-infected MDA5 and TLR3 null mice showed reduced lung inflammatory responses and reduced airways responsiveness. Finally, RV-infected MDA5 null mice with allergic airways disease showed lower viral titers despite deficient IFN responses, and allergic MDA5 and TLR3 null mice each showed decreased RV-induced airway inflammatory and contractile responses. These results suggest that, in the context of RV infection, binding of viral dsRNA to MDA5 and TLR3 initiates pro-inflammatory signaling pathways leading to airways inflammation and hyperresponsiveness.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Role of double-stranded RNA pattern recognition receptors in rhinovirus-induced airway epithelial cell responses.J Immunol. 2009 Dec 1;183(11):6989-97. doi: 10.4049/jimmunol.0901386. Epub 2009 Nov 4. J Immunol. 2009. PMID: 19890046 Free PMC article.
-
TLR3 and MDA5 signalling, although not expression, is impaired in asthmatic epithelial cells in response to rhinovirus infection.Clin Exp Allergy. 2014 Jan;44(1):91-101. doi: 10.1111/cea.12218. Clin Exp Allergy. 2014. PMID: 24131248
-
Co-ordinated role of TLR3, RIG-I and MDA5 in the innate response to rhinovirus in bronchial epithelium.PLoS Pathog. 2010 Nov 4;6(11):e1001178. doi: 10.1371/journal.ppat.1001178. PLoS Pathog. 2010. PMID: 21079690 Free PMC article.
-
MDA5-filament, dynamics and disease.Curr Opin Virol. 2015 Jun;12:20-5. doi: 10.1016/j.coviro.2015.01.011. Epub 2015 Feb 9. Curr Opin Virol. 2015. PMID: 25676875 Free PMC article. Review.
-
TLR7/9 versus TLR3/MDA5 signaling during virus infections and diabetes.J Leukoc Biol. 2011 Oct;90(4):691-701. doi: 10.1189/jlb.0311166. Epub 2011 Aug 15. J Leukoc Biol. 2011. PMID: 21844166 Free PMC article. Review.
Cited by
-
Nasal cytokine responses to natural colds in asthmatic children.Clin Exp Allergy. 2012 Dec;42(12):1734-44. doi: 10.1111/cea.12005. Clin Exp Allergy. 2012. PMID: 23181789 Free PMC article.
-
Rhinovirus-Induced Airway Disease: A Model to Understand the Antiviral and Th2 Epithelial Immune Dysregulation in Childhood Asthma.J Investig Med. 2015 Aug;63(6):792-5. doi: 10.1097/JIM.0000000000000209. J Investig Med. 2015. PMID: 26057561 Free PMC article. Review.
-
Toll-like receptor 3 in viral pathogenesis: friend or foe?Immunology. 2013 Oct;140(2):153-67. doi: 10.1111/imm.12143. Immunology. 2013. PMID: 23909285 Free PMC article. Review.
-
Distinct roles for MDA5 and TLR3 in the acute response to inhaled double-stranded RNA.PLoS One. 2019 May 8;14(5):e0216056. doi: 10.1371/journal.pone.0216056. eCollection 2019. PLoS One. 2019. PMID: 31067281 Free PMC article.
-
Rhinovirus infection induces interleukin-13 production from CD11b-positive, M2-polarized exudative macrophages.Am J Respir Cell Mol Biol. 2015 Feb;52(2):205-16. doi: 10.1165/rcmb.2014-0068OC. Am J Respir Cell Mol Biol. 2015. PMID: 25029349 Free PMC article.
References
-
- Doyle SE, Schreckhise H, Khuu-Duong K, Henderson K, Rosler R, et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology. 2006;44:896–906. - PubMed
-
- Hornung V, Ellegast J, Kim SK, Brzozka K, Jung A, et al. 5′-Triphosphate RNA is the ligand for RIG-I. Science. 2006;314:994–997. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
