

AAV exploits subcellular stress associated with inflammation, endoplasmic reticulum expansion, and misfolded proteins in models of cystic fibrosis
- PMID: 21625534
- PMCID: PMC3098238
- DOI: 10.1371/journal.ppat.1002053
AAV exploits subcellular stress associated with inflammation, endoplasmic reticulum expansion, and misfolded proteins in models of cystic fibrosis
Abstract
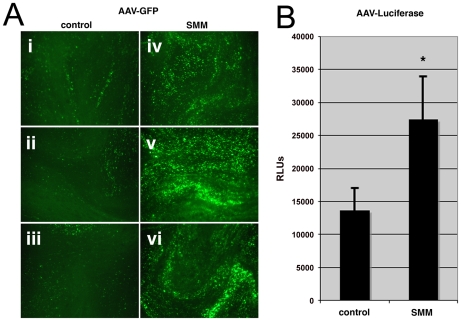
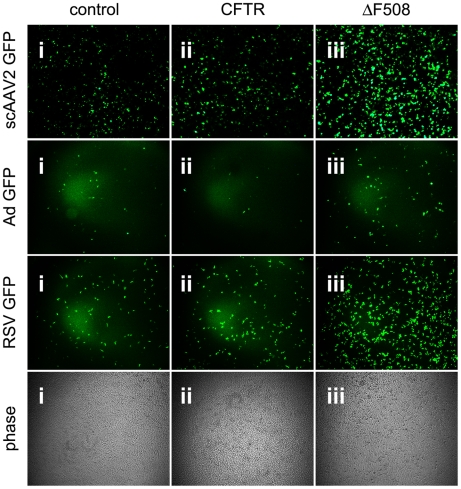
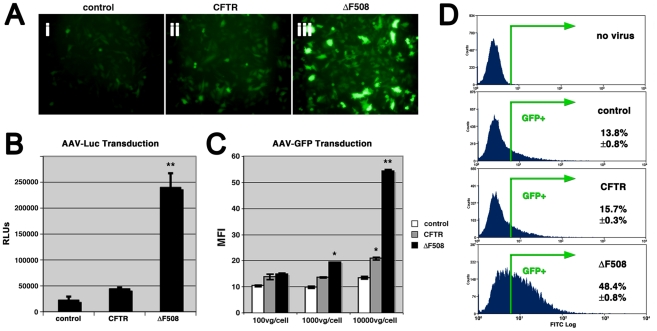
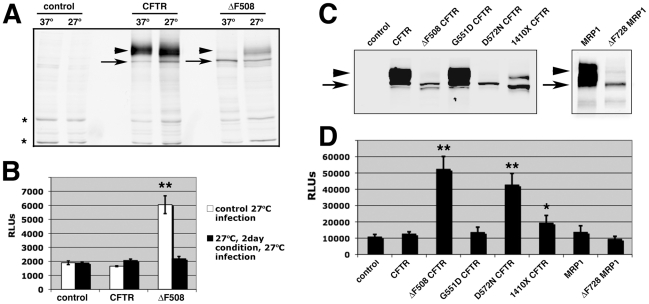
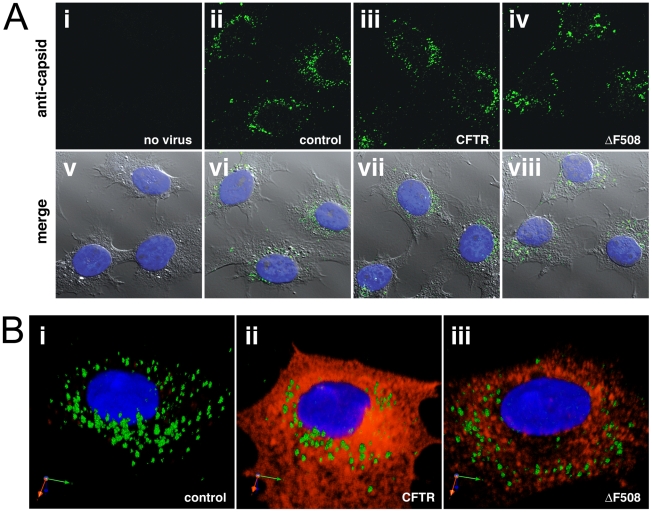
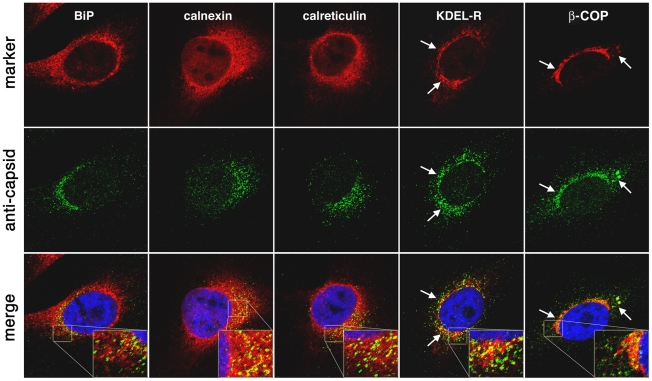
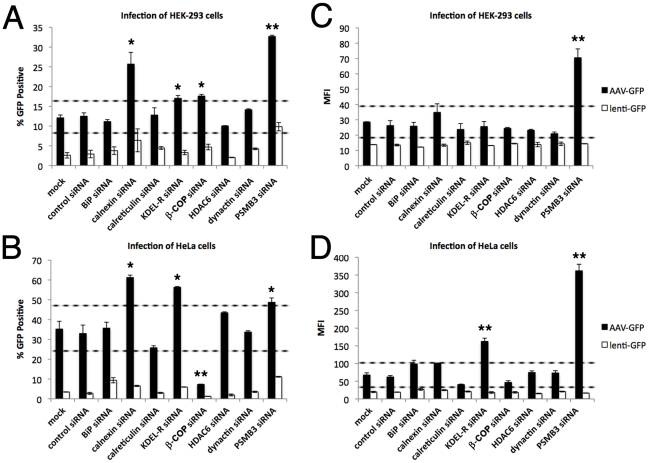
Barriers to infection act at multiple levels to prevent viruses, bacteria, and parasites from commandeering host cells for their own purposes. An intriguing hypothesis is that if a cell experiences stress, such as that elicited by inflammation, endoplasmic reticulum (ER) expansion, or misfolded proteins, then subcellular barriers will be less effective at preventing viral infection. Here we have used models of cystic fibrosis (CF) to test whether subcellular stress increases susceptibility to adeno-associated virus (AAV) infection. In human airway epithelium cultured at an air/liquid interface, physiological conditions of subcellular stress and ER expansion were mimicked using supernatant from mucopurulent material derived from CF lungs. Using this inflammatory stimulus to recapitulate stress found in diseased airways, we demonstrated that AAV infection was significantly enhanced. Since over 90% of CF cases are associated with a misfolded variant of Cystic Fibrosis Transmembrane Conductance Regulator (ΔF508-CFTR), we then explored whether the presence of misfolded proteins could independently increase susceptibility to AAV infection. In these models, AAV was an order of magnitude more efficient at transducing cells expressing ΔF508-CFTR than in cells expressing wild-type CFTR. Rescue of misfolded ΔF508-CFTR under low temperature conditions restored viral transduction efficiency to that demonstrated in controls, suggesting effects related to protein misfolding were responsible for increasing susceptibility to infection. By testing other CFTR mutants, G551D, D572N, and 1410X, we have shown this phenomenon is common to other misfolded proteins and not related to loss of CFTR activity. The presence of misfolded proteins did not affect cell surface attachment of virus or influence expression levels from promoter transgene cassettes in plasmid transfection studies, indicating exploitation occurs at the level of virion trafficking or processing. Thus, we surmised that factors enlisted to process misfolded proteins such as ΔF508-CFTR in the secretory pathway also act to restrict viral infection. In line with this hypothesis, we found that AAV trafficked to the microtubule organizing center and localized near Golgi/ER transport proteins. Moreover, AAV infection efficiency could be modulated with siRNA-mediated knockdown of proteins involved in processing ΔF508-CFTR or sorting retrograde cargo from the Golgi and ER (calnexin, KDEL-R, β-COP, and PSMB3). In summary, our data support a model where AAV exploits a compromised secretory system and, importantly, underscore the gravity with which a stressed subcellular environment, under internal or external insults, can impact infection efficiency.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Human heat shock protein 105/110 kDa (Hsp105/110) regulates biogenesis and quality control of misfolded cystic fibrosis transmembrane conductance regulator at multiple levels.J Biol Chem. 2012 Jun 1;287(23):19158-70. doi: 10.1074/jbc.M111.297580. Epub 2012 Apr 13. J Biol Chem. 2012. PMID: 22505710 Free PMC article.
-
Cystic fibrosis airway epithelial Ca2+ i signaling: the mechanism for the larger agonist-mediated Ca2+ i signals in human cystic fibrosis airway epithelia.J Biol Chem. 2005 Mar 18;280(11):10202-9. doi: 10.1074/jbc.M410617200. Epub 2005 Jan 12. J Biol Chem. 2005. PMID: 15647273
-
Base treatment corrects defects due to misfolding of mutant cystic fibrosis transmembrane conductance regulator.Gastroenterology. 2005 Dec;129(6):1979-90. doi: 10.1053/j.gastro.2005.08.049. Gastroenterology. 2005. PMID: 16344066
-
Control of cystic fibrosis transmembrane conductance regulator membrane trafficking: not just from the endoplasmic reticulum to the Golgi.FEBS J. 2013 Sep;280(18):4396-406. doi: 10.1111/febs.12392. Epub 2013 Jul 5. FEBS J. 2013. PMID: 23773658 Review.
-
Protein processing and inflammatory signaling in Cystic Fibrosis: challenges and therapeutic strategies.Curr Mol Med. 2010 Feb;10(1):82-94. doi: 10.2174/156652410791065408. Curr Mol Med. 2010. PMID: 20205681 Free PMC article. Review.
Cited by
-
AAV Immunotoxicity: Implications in Anti-HBV Gene Therapy.Microorganisms. 2023 Dec 14;11(12):2985. doi: 10.3390/microorganisms11122985. Microorganisms. 2023. PMID: 38138129 Free PMC article. Review.
-
Nanotechnology approaches for inhalation treatment of fibrosis.J Drug Target. 2013 Dec;21(10):914-25. doi: 10.3109/1061186X.2013.829078. Epub 2013 Aug 27. J Drug Target. 2013. PMID: 23978292 Free PMC article. Review.
-
Syntaxin 5-dependent retrograde transport to the trans-Golgi network is required for adeno-associated virus transduction.J Virol. 2015 Feb;89(3):1673-87. doi: 10.1128/JVI.02520-14. Epub 2014 Nov 19. J Virol. 2015. PMID: 25410859 Free PMC article.
-
Cystic fibrosis: a mucosal immunodeficiency syndrome.Nat Med. 2012 Apr 5;18(4):509-19. doi: 10.1038/nm.2715. Nat Med. 2012. PMID: 22481418 Free PMC article. Review.
-
Arsenic trioxide stabilizes accumulations of adeno-associated virus virions at the perinuclear region, increasing transduction in vitro and in vivo.J Virol. 2013 Apr;87(8):4571-83. doi: 10.1128/JVI.03443-12. Epub 2013 Feb 13. J Virol. 2013. PMID: 23408604 Free PMC article.
References
-
- O'Sullivan BP, Freedman SD. Cystic fibrosis. Lancet. 2009;373:1891–1904. - PubMed
-
- Sferra TJ, Collins FS. The molecular biology of cystic fibrosis. Annu Rev Med. 1993;44:133–144. - PubMed
-
- Qu BH, Strickland EH, Thomas PJ. Localization and suppression of a kinetic defect in cystic fibrosis transmembrane conductance regulator folding. J Biol Chem. 1997;272:15739–15744. - PubMed
-
- Riordan JR. Assembly of functional CFTR chloride channels. Annu Rev Physiol. 2005;67:701–718. - PubMed
-
- Gelman MS, Kannegaard ES, Kopito RR. A principal role for the proteasome in endoplasmic reticulum-associated degradation of misfolded intracellular cystic fibrosis transmembrane conductance regulator. J Biol Chem. 2002;277:11709–11714. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
