

Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth
- PMID: 21620777
- PMCID: PMC3157296
- DOI: 10.1016/j.ccr.2011.05.006
Cell autonomous role of PTEN in regulating castration-resistant prostate cancer growth
Abstract
Alteration of the PTEN/PI3K pathway is associated with late-stage and castrate-resistant prostate cancer (CRPC). However, how PTEN loss is involved in CRPC development is not clear. Here, we show that castration-resistant growth is an intrinsic property of Pten null prostate cancer (CaP) cells, independent of cancer development stage. PTEN loss suppresses androgen-responsive gene expressions by modulating androgen receptor (AR) transcription factor activity. Conditional deletion of Ar in the epithelium promotes the proliferation of Pten null cancer cells, at least in part, by downregulating the androgen-responsive gene Fkbp5 and preventing PHLPP-mediated AKT inhibition. Our findings identify PI3K and AR pathway crosstalk as a mechanism of CRPC development, with potentially important implications for CaP etiology and therapy.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures

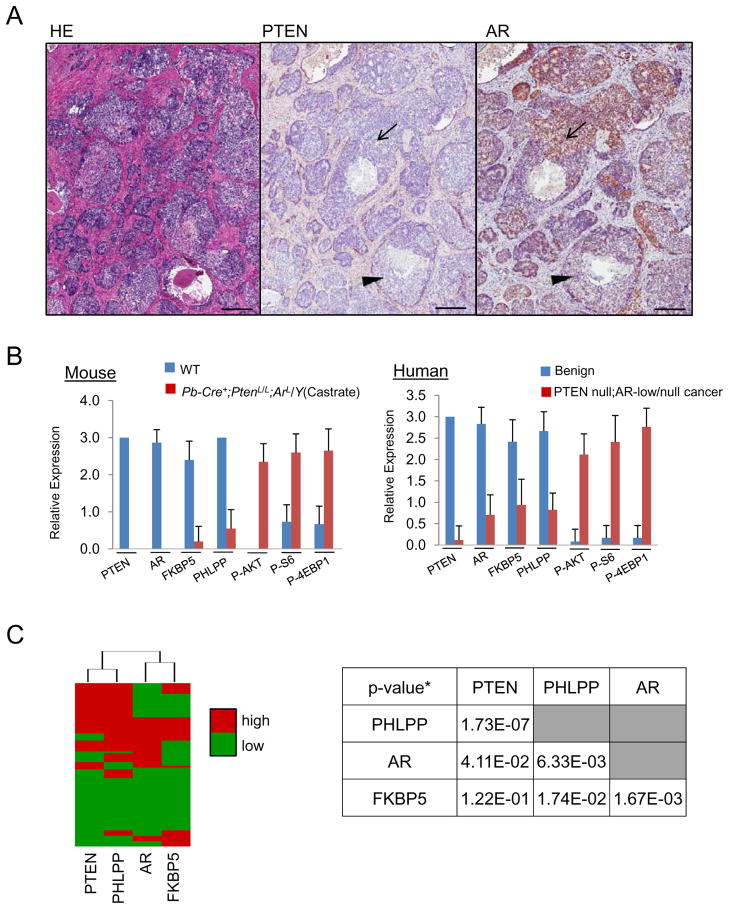
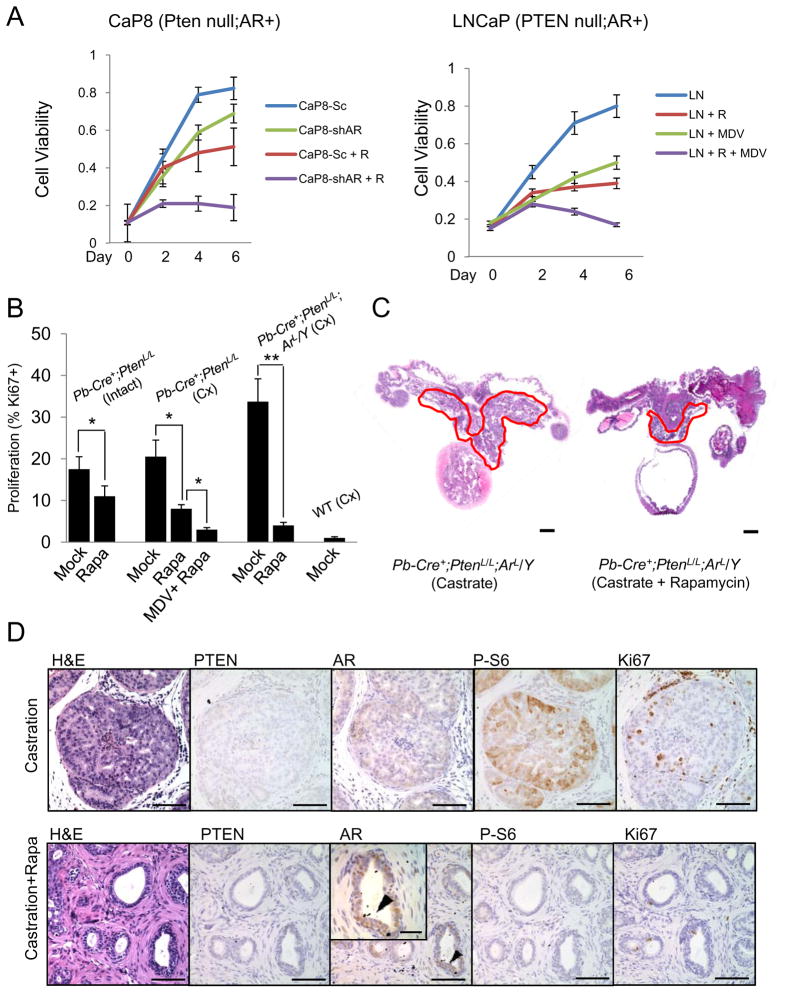
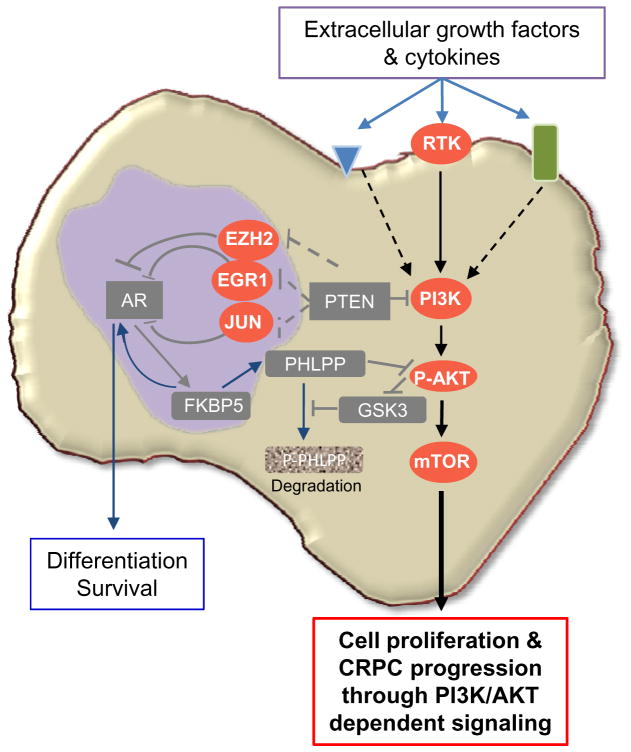
Comment in
-
Prostate cancer: loss of PTEN promotes progression of prostate cancer in an androgen-independent manner.Nat Rev Urol. 2011 Aug 10;8(8):412. doi: 10.1038/nrurol.2011.109. Nat Rev Urol. 2011. PMID: 21829237 No abstract available.
Similar articles
-
Effect of dietary polyunsaturated fatty acids on castration-resistant Pten-null prostate cancer.Carcinogenesis. 2012 Feb;33(2):404-12. doi: 10.1093/carcin/bgr290. Epub 2011 Dec 8. Carcinogenesis. 2012. PMID: 22159221 Free PMC article.
-
The steroid receptor coactivator-3 is required for the development of castration-resistant prostate cancer.Cancer Res. 2013 Jul 1;73(13):3997-4008. doi: 10.1158/0008-5472.CAN-12-3929. Epub 2013 May 6. Cancer Res. 2013. PMID: 23650284 Free PMC article.
-
Resveratrol regulates the PTEN/AKT pathway through androgen receptor-dependent and -independent mechanisms in prostate cancer cell lines.Hum Mol Genet. 2010 Nov 15;19(22):4319-29. doi: 10.1093/hmg/ddq354. Epub 2010 Aug 20. Hum Mol Genet. 2010. PMID: 20729295 Free PMC article.
-
Interplay Among PI3K/AKT, PTEN/FOXO and AR Signaling in Prostate Cancer.Adv Exp Med Biol. 2019;1210:319-331. doi: 10.1007/978-3-030-32656-2_14. Adv Exp Med Biol. 2019. PMID: 31900915 Review.
-
Deregulated PTEN/PI3K/AKT/mTOR signaling in prostate cancer: Still a potential druggable target?Biochim Biophys Acta Mol Cell Res. 2020 Sep;1867(9):118731. doi: 10.1016/j.bbamcr.2020.118731. Epub 2020 Apr 29. Biochim Biophys Acta Mol Cell Res. 2020. PMID: 32360668 Review.
Cited by
-
Circ_0001047 inhibits prostate cancer progression and enhances abiraterone sensitivity via miR-122-5p/FKBP5/PHLPP1/AKT axis in vitro.Discov Oncol. 2024 Oct 17;15(1):569. doi: 10.1007/s12672-024-01408-z. Discov Oncol. 2024. PMID: 39419900 Free PMC article.
-
Pleckstrin-2 Mediates the Activation of AKT in Prostate Cancer and Is Repressed by Androgen Receptor.Am J Pathol. 2024 Oct;194(10):1986-1996. doi: 10.1016/j.ajpath.2024.07.004. Epub 2024 Jul 26. Am J Pathol. 2024. PMID: 39069167
-
GPCR-Gα13 Involvement in Mitochondrial Function, Oxidative Stress, and Prostate Cancer.Int J Mol Sci. 2024 Jun 28;25(13):7162. doi: 10.3390/ijms25137162. Int J Mol Sci. 2024. PMID: 39000269 Free PMC article. Review.
-
Metabolic adaptations in prostate cancer.Br J Cancer. 2024 Nov;131(8):1250-1262. doi: 10.1038/s41416-024-02762-z. Epub 2024 Jul 5. Br J Cancer. 2024. PMID: 38969865 Free PMC article. Review.
-
Androgen deprivation therapy exacerbates Alzheimer's-associated cognitive decline via increased brain immune cell infiltration.Sci Adv. 2024 Jun 21;10(25):eadn8709. doi: 10.1126/sciadv.adn8709. Epub 2024 Jun 21. Sci Adv. 2024. PMID: 38905345 Free PMC article.
References
-
- Abbas F, Civantos F, Benedetto P, Soloway MS. Small cell carcinoma of the bladder and prostate. Urology. 1995;46:617–630. - PubMed
-
- Abdulkadir SA, Qu Z, Garabedian E, Song SK, Peters TJ, Svaren J, Carbone JM, Naughton CK, Catalona WJ, Ackerman JJ, et al. Impaired prostate tumorigenesis in Egr1-deficient mice. Nat Med. 2001;7:101–107. - PubMed
-
- Attard G, Cooper CS, de Bono JS. Steroid hormone receptors in prostate cancer: a hard habit to break? Cancer Cell. 2009a;16:458–462. - PubMed
-
- Attard G, Reid AH, Olmos D, de Bono JS. Antitumor activity with CYP17 blockade indicates that castration-resistant prostate cancer frequently remains hormone driven. Cancer Res. 2009b;69:4937–4940. - PubMed
-
- Brognard J, Sierecki E, Gao T, Newton AC. PHLPP and a second isoform, PHLPP2, differentially attenuate the amplitude of Akt signaling by regulating distinct Akt isoforms. Mol Cell. 2007;25:917–931. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
