

MicroRNA sequence and expression analysis in breast tumors by deep sequencing
- PMID: 21586611
- PMCID: PMC3129492
- DOI: 10.1158/0008-5472.CAN-11-0608
MicroRNA sequence and expression analysis in breast tumors by deep sequencing
Abstract
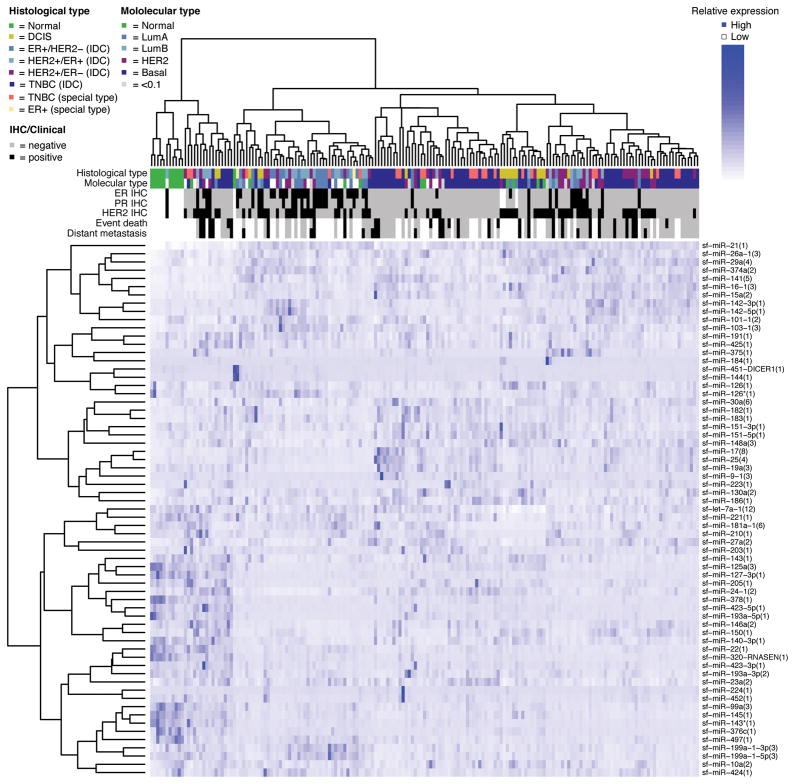
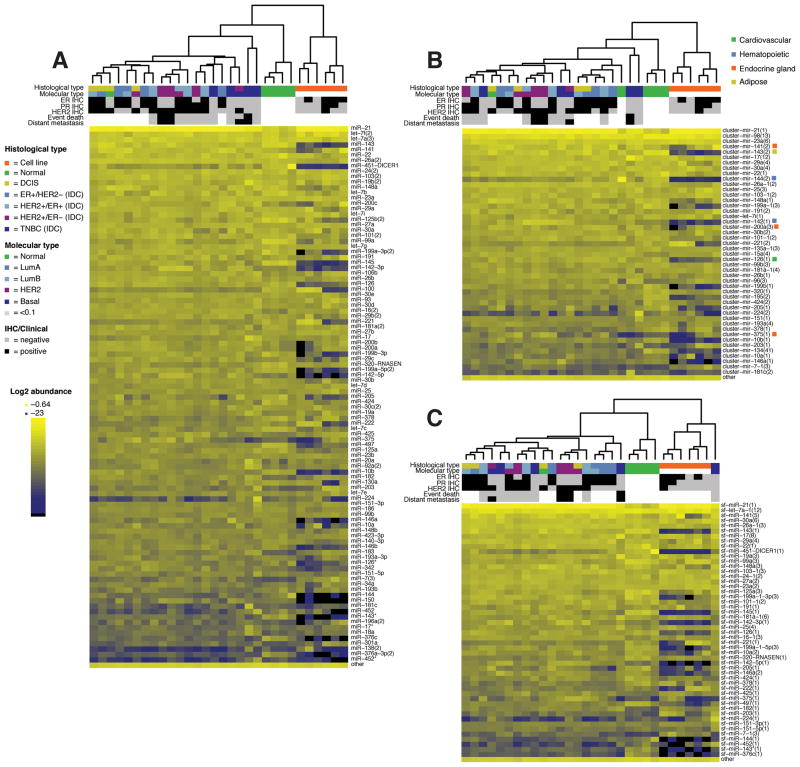
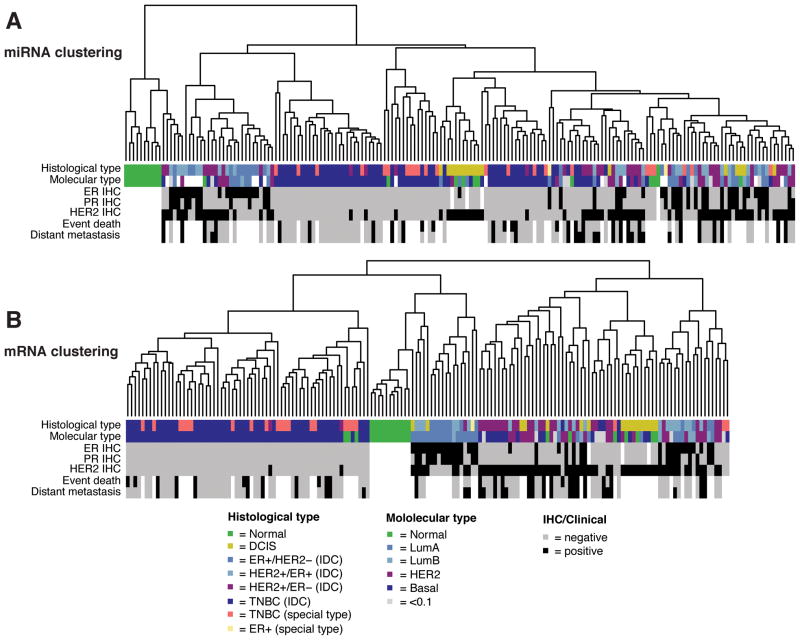
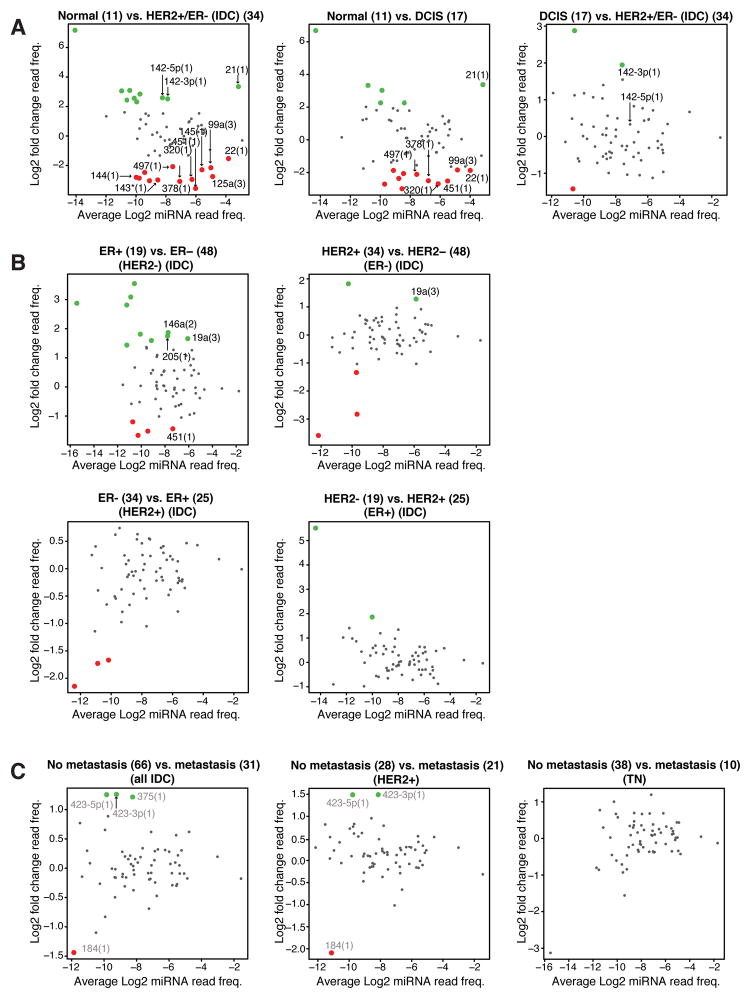
MicroRNAs (miRNA) regulate many genes critical for tumorigenesis. We profiled miRNAs from 11 normal breast tissues, 17 noninvasive, 151 invasive breast carcinomas, and 6 cell lines by in-house-developed barcoded Solexa sequencing. miRNAs were organized in genomic clusters representing promoter-controlled miRNA expression and sequence families representing seed sequence-dependent miRNA target regulation. Unsupervised clustering of samples by miRNA sequence families best reflected the clustering based on mRNA expression available for this sample set. Clustering and comparative analysis of miRNA read frequencies showed that normal breast samples were separated from most noninvasive ductal carcinoma in situ and invasive carcinomas by increased miR-21 (the most abundant miRNA in carcinomas) and multiple decreased miRNA families (including miR-98/let-7), with most miRNA changes apparent already in the noninvasive carcinomas. In addition, patients that went on to develop metastasis showed increased expression of mir-423, and triple-negative breast carcinomas were most distinct from other tumor subtypes due to upregulation of the mir~17-92 cluster. However, absolute miRNA levels between normal breast and carcinomas did not reveal any significant differences. We also discovered two polymorphic nucleotide variations among the more abundant miRNAs miR-181a (T19G) and miR-185 (T16G), but we did not identify nucleotide variations expected for classical tumor suppressor function associated with miRNAs. The differentiation of tumor subtypes and prediction of metastasis based on miRNA levels is statistically possible but is not driven by deregulation of abundant miRNAs, implicating far fewer miRNAs in tumorigenic processes than previously suggested.
©2011 AACR.
Conflict of interest statement
T. T. is cofounder and scientific advisor to Alnylam Pharmaceuticals and scientific advisor to Regulus Therapeutics.
Figures
Similar articles
-
DNA methylation contributes to deregulation of 12 cancer-associated microRNAs and breast cancer progression.Gene. 2017 Mar 10;604:1-8. doi: 10.1016/j.gene.2016.12.018. Epub 2016 Dec 18. Gene. 2017. PMID: 27998789
-
miRNA expression patterns in normal breast tissue and invasive breast cancers of BRCA1 and BRCA2 germ-line mutation carriers.Oncotarget. 2015 Oct 13;6(31):32115-37. doi: 10.18632/oncotarget.5617. Oncotarget. 2015. PMID: 26378051 Free PMC article.
-
Role of deregulated microRNAs in breast cancer progression using FFPE tissue.PLoS One. 2013;8(1):e54213. doi: 10.1371/journal.pone.0054213. Epub 2013 Jan 23. PLoS One. 2013. PMID: 23372687 Free PMC article.
-
miRNA expression in breast cancer varies with lymph node metastasis and other clinicopathologic features.IUBMB Life. 2014 May;66(5):371-7. doi: 10.1002/iub.1273. Epub 2014 May 20. IUBMB Life. 2014. PMID: 24846313
-
From microRNA functions to microRNA therapeutics: novel targets and novel drugs in breast cancer research and treatment (Review).Int J Oncol. 2013 Oct;43(4):985-94. doi: 10.3892/ijo.2013.2059. Epub 2013 Aug 12. Int J Oncol. 2013. PMID: 23939688 Free PMC article. Review.
Cited by
-
Comparative studies of two methods for miRNA isolation from milk whey.J Zhejiang Univ Sci B. 2015 Jun;16(6):533-40. doi: 10.1631/jzus.B1400355. J Zhejiang Univ Sci B. 2015. PMID: 26055915 Free PMC article.
-
MicroRNAs in human cancer.Adv Exp Med Biol. 2013;774:1-20. doi: 10.1007/978-94-007-5590-1_1. Adv Exp Med Biol. 2013. PMID: 23377965 Free PMC article. Review.
-
MicroRNA-509-3p Inhibits Cancer Cell Proliferation and Migration via Upregulation of XIAP in Gastric Cancer Cells.Oncol Res. 2017 Mar 13;25(3):455-461. doi: 10.3727/096504016X14747283032017. Epub 2016 Sep 30. Oncol Res. 2017. PMID: 27697095 Free PMC article.
-
BioGraph: a web application and a graph database for querying and analyzing bioinformatics resources.BMC Syst Biol. 2018 Nov 20;12(Suppl 5):98. doi: 10.1186/s12918-018-0616-4. BMC Syst Biol. 2018. PMID: 30458802 Free PMC article.
-
High-throughput sequencing analysis of differentially expressed miRNAs and target genes in ischemia/reperfusion injury and apelin-13 neuroprotection.Neural Regen Res. 2018 Feb;13(2):265-271. doi: 10.4103/1673-5374.226397. Neural Regen Res. 2018. PMID: 29557376 Free PMC article.
References
-
- van de Vijver MJ, He YD, van't Veer LJ, et al. A gene-expression signature as a predictor of survival in breast cancer. N Engl J Med. 2002;347(25):1999–2009. - PubMed
-
- Perou CM, Sorlie T, Eisen MB, et al. Molecular portraits of human breast tumours. Nature. 2000;406(6797):747–52. - PubMed
-
- Fabian MR, Sonenberg N, Filipowicz W. Regulation of mRNA translation and stability by microRNAs. Annu Rev Biochem. 2010;79:351–79. - PubMed
-
- Krol J, Loedige I, Filipowicz W. The widespread regulation of microRNA biogenesis, function and decay. Nat Rev Genet. 2010;11(9):597–610. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
