

A hierarchical whole-body modeling approach elucidates the link between in Vitro insulin signaling and in Vivo glucose homeostasis
- PMID: 21572040
- PMCID: PMC3138269
- DOI: 10.1074/jbc.M110.188987
A hierarchical whole-body modeling approach elucidates the link between in Vitro insulin signaling and in Vivo glucose homeostasis
Abstract
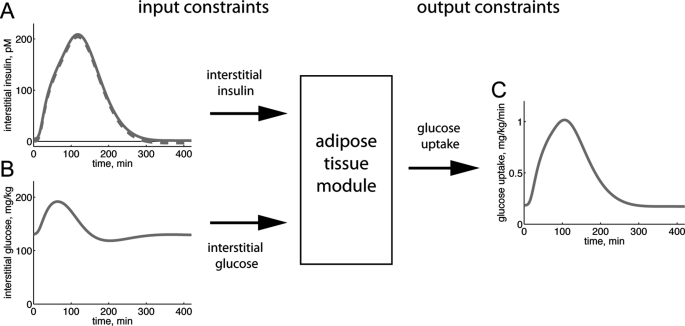
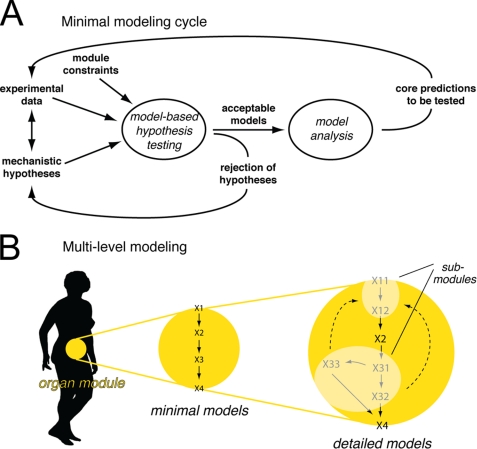
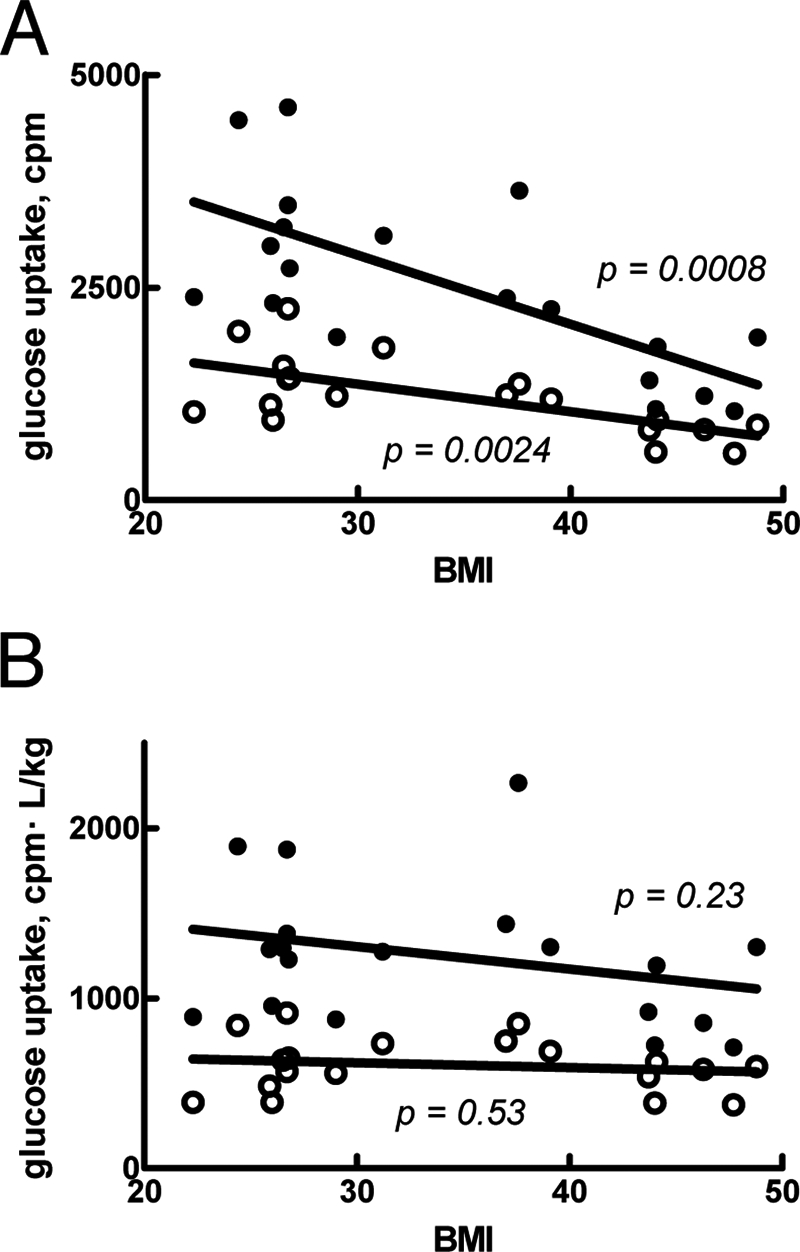
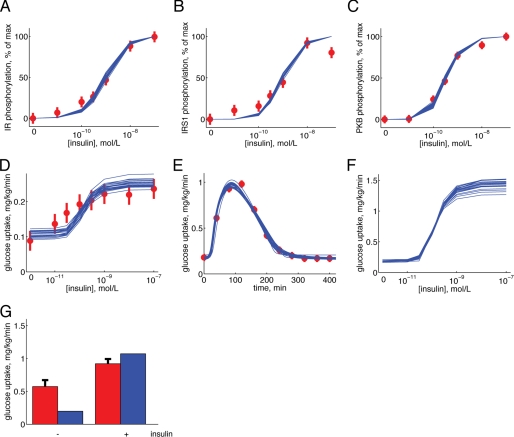
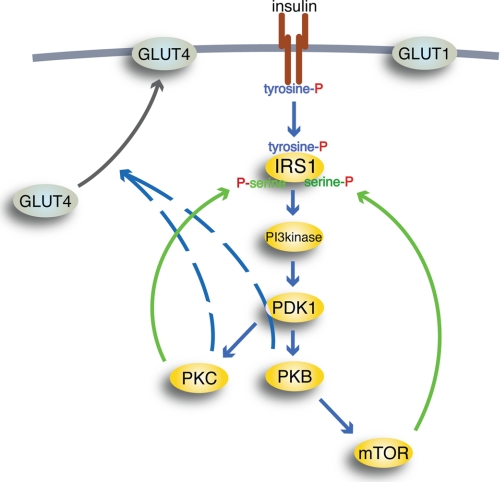
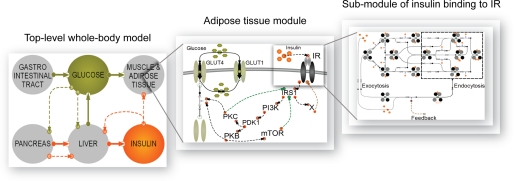
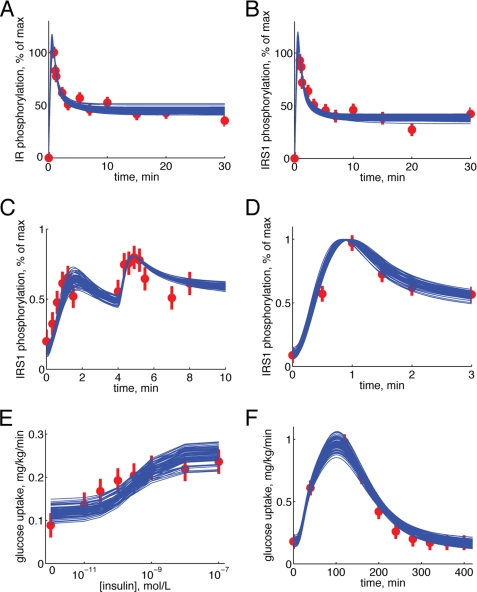
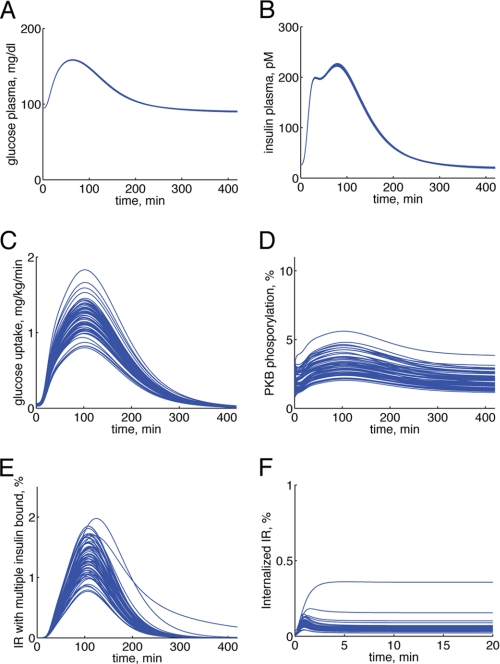
Type 2 diabetes is a metabolic disease that profoundly affects energy homeostasis. The disease involves failure at several levels and subsystems and is characterized by insulin resistance in target cells and tissues (i.e. by impaired intracellular insulin signaling). We have previously used an iterative experimental-theoretical approach to unravel the early insulin signaling events in primary human adipocytes. That study, like most insulin signaling studies, is based on in vitro experimental examination of cells, and the in vivo relevance of such studies for human beings has not been systematically examined. Herein, we develop a hierarchical model of the adipose tissue, which links intracellular insulin control of glucose transport in human primary adipocytes with whole-body glucose homeostasis. An iterative approach between experiments and minimal modeling allowed us to conclude that it is not possible to scale up the experimentally determined glucose uptake by the isolated adipocytes to match the glucose uptake profile of the adipose tissue in vivo. However, a model that additionally includes insulin effects on blood flow in the adipose tissue and GLUT4 translocation due to cell handling can explain all data, but neither of these additions is sufficient independently. We also extend the minimal model to include hierarchical dynamic links to more detailed models (both to our own models and to those by others), which act as submodules that can be turned on or off. The resulting multilevel hierarchical model can merge detailed results on different subsystems into a coherent understanding of whole-body glucose homeostasis. This hierarchical modeling can potentially create bridges between other experimental model systems and the in vivo human situation and offers a framework for systematic evaluation of the physiological relevance of in vitro obtained molecular/cellular experimental data.
Figures

Similar articles
-
GLUT4 defects in adipose tissue are early signs of metabolic alterations in Alms1GT/GT, a mouse model for obesity and insulin resistance.PLoS One. 2014 Oct 9;9(10):e109540. doi: 10.1371/journal.pone.0109540. eCollection 2014. PLoS One. 2014. PMID: 25299671 Free PMC article.
-
Deletion of Rab GAP AS160 modifies glucose uptake and GLUT4 translocation in primary skeletal muscles and adipocytes and impairs glucose homeostasis.Am J Physiol Endocrinol Metab. 2012 Nov 15;303(10):E1273-86. doi: 10.1152/ajpendo.00316.2012. Epub 2012 Sep 25. Am J Physiol Endocrinol Metab. 2012. PMID: 23011063 Free PMC article.
-
Defective insulin-stimulated GLUT4 translocation in brown adipocytes induces systemic glucose homeostasis dysregulation independent of thermogenesis in female mice.Mol Metab. 2021 Nov;53:101305. doi: 10.1016/j.molmet.2021.101305. Epub 2021 Jul 21. Mol Metab. 2021. PMID: 34303022 Free PMC article.
-
The brown adipose cell: a unique model for understanding the molecular mechanism of insulin resistance.Mini Rev Med Chem. 2005 Mar;5(3):269-78. doi: 10.2174/1389557053175380. Mini Rev Med Chem. 2005. PMID: 15777261 Review.
-
Intracellular organization of insulin signaling and GLUT4 translocation.Recent Prog Horm Res. 2001;56:175-93. doi: 10.1210/rp.56.1.175. Recent Prog Horm Res. 2001. PMID: 11237212 Review.
Cited by
-
An integrated strategy for prediction uncertainty analysis.Bioinformatics. 2012 Apr 15;28(8):1130-5. doi: 10.1093/bioinformatics/bts088. Epub 2012 Feb 21. Bioinformatics. 2012. PMID: 22355081 Free PMC article.
-
Computationally Modeling Lipid Metabolism and Aging: A Mini-review.Comput Struct Biotechnol J. 2014 Nov 15;13:38-46. doi: 10.1016/j.csbj.2014.11.006. eCollection 2015. Comput Struct Biotechnol J. 2014. PMID: 25750699 Free PMC article. Review.
-
Insulin Signaling in Insulin Resistance States and Cancer: A Modeling Analysis.PLoS One. 2016 May 5;11(5):e0154415. doi: 10.1371/journal.pone.0154415. eCollection 2016. PLoS One. 2016. PMID: 27149630 Free PMC article.
-
Evaluating systems pharmacology models is different from evaluating standard pharmacokinetic-pharmacodynamic models.CPT Pharmacometrics Syst Pharmacol. 2014 Feb 19;3(2):e101. doi: 10.1038/psp.2013.77. CPT Pharmacometrics Syst Pharmacol. 2014. PMID: 24552986 Free PMC article.
-
Translational Quantitative Systems Pharmacology in Drug Development: from Current Landscape to Good Practices.AAPS J. 2019 Jun 3;21(4):72. doi: 10.1208/s12248-019-0339-5. AAPS J. 2019. PMID: 31161268 Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
