

A user's guide to the encyclopedia of DNA elements (ENCODE)
- PMID: 21526222
- PMCID: PMC3079585
- DOI: 10.1371/journal.pbio.1001046
A user's guide to the encyclopedia of DNA elements (ENCODE)
Abstract
The mission of the Encyclopedia of DNA Elements (ENCODE) Project is to enable the scientific and medical communities to interpret the human genome sequence and apply it to understand human biology and improve health. The ENCODE Consortium is integrating multiple technologies and approaches in a collective effort to discover and define the functional elements encoded in the human genome, including genes, transcripts, and transcriptional regulatory regions, together with their attendant chromatin states and DNA methylation patterns. In the process, standards to ensure high-quality data have been implemented, and novel algorithms have been developed to facilitate analysis. Data and derived results are made available through a freely accessible database. Here we provide an overview of the project and the resources it is generating and illustrate the application of ENCODE data to interpret the human genome.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures

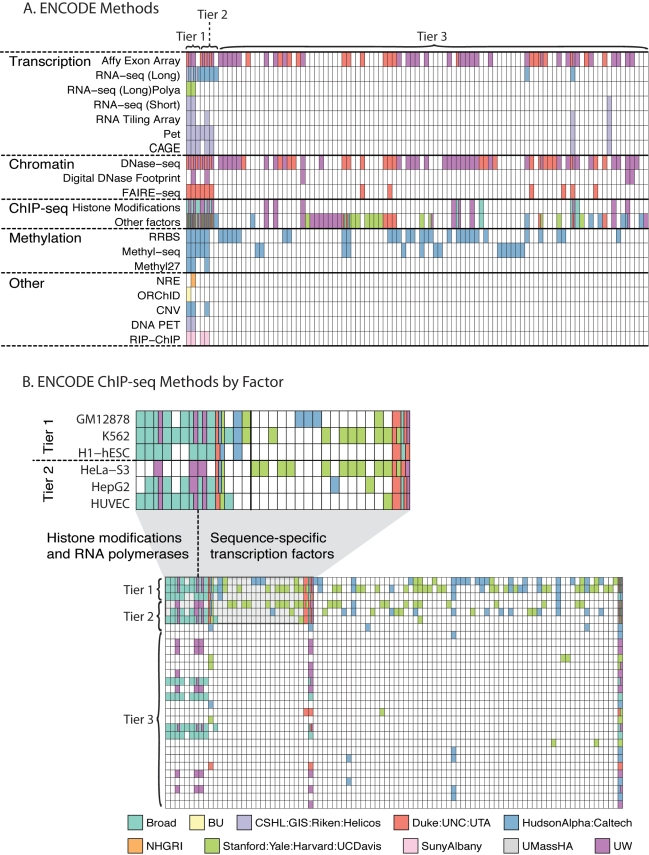

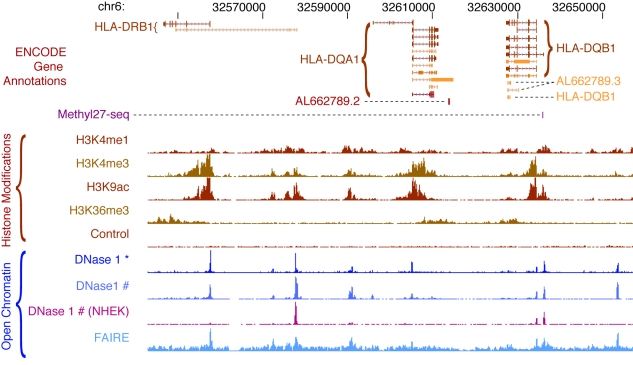




Similar articles
-
The ENCODE (ENCyclopedia Of DNA Elements) Project.Science. 2004 Oct 22;306(5696):636-40. doi: 10.1126/science.1105136. Science. 2004. PMID: 15499007
-
The ENCODE Portal as an Epigenomics Resource.Curr Protoc Bioinformatics. 2019 Dec;68(1):e89. doi: 10.1002/cpbi.89. Curr Protoc Bioinformatics. 2019. PMID: 31751002 Free PMC article.
-
Expanded encyclopaedias of DNA elements in the human and mouse genomes.Nature. 2020 Jul;583(7818):699-710. doi: 10.1038/s41586-020-2493-4. Epub 2020 Jul 29. Nature. 2020. PMID: 32728249 Free PMC article.
-
A brief review on the Human Encyclopedia of DNA Elements (ENCODE) project.Genomics Proteomics Bioinformatics. 2013 Jun;11(3):135-41. doi: 10.1016/j.gpb.2013.05.001. Epub 2013 May 28. Genomics Proteomics Bioinformatics. 2013. PMID: 23722115 Free PMC article. Review.
-
Epigenetics, chromatin and genome organization: recent advances from the ENCODE project.J Intern Med. 2014 Sep;276(3):201-14. doi: 10.1111/joim.12231. Epub 2014 Mar 27. J Intern Med. 2014. PMID: 24605849 Review.
Cited by
-
Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations.Nat Genet. 2015 Sep;47(9):979-986. doi: 10.1038/ng.3359. Epub 2015 Jul 20. Nat Genet. 2015. PMID: 26192919 Free PMC article.
-
Gene co-expression and histone modification signatures are associated with melanoma progression, epithelial-to-mesenchymal transition, and metastasis.Clin Epigenetics. 2020 Aug 24;12(1):127. doi: 10.1186/s13148-020-00910-9. Clin Epigenetics. 2020. PMID: 32831131 Free PMC article.
-
Functional characterization of motif sequences under purifying selection.Nucleic Acids Res. 2013 Feb 1;41(4):2105-20. doi: 10.1093/nar/gks1456. Epub 2013 Jan 8. Nucleic Acids Res. 2013. PMID: 23303791 Free PMC article.
-
DOT1L promotes angiogenesis through cooperative regulation of VEGFR2 with ETS-1.Oncotarget. 2016 Oct 25;7(43):69674-69687. doi: 10.18632/oncotarget.11939. Oncotarget. 2016. PMID: 27626484 Free PMC article.
-
Identification of recurrent NAB2-STAT6 gene fusions in solitary fibrous tumor by integrative sequencing.Nat Genet. 2013 Feb;45(2):180-5. doi: 10.1038/ng.2509. Epub 2013 Jan 13. Nat Genet. 2013. PMID: 23313952 Free PMC article.
References
-
- Collins F. S, Green E. D, Guttmacher A. E, Guyer M. S. A vision for the future of genomics research. Nature. 2003;422:835–847. - PubMed
-
- ENCODE Project Consortium. The ENCODE (ENCyclopedia Of DNA Elements) Project. Science. 2004;306:636–640. - PubMed
-
- Mouse Genome Sequencing Consortium. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. - PubMed
-
- Chiaromonte F, Weber R. J, Roskin K. M, Diekhans M, Kent W. J, Haussler D. The share of human genomic DNA under selection estimated from human-mouse genomic alignments. Cold Spring Harb Symp Quant Biol. 2003;68:245–254. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
