

MDAnalysis: a toolkit for the analysis of molecular dynamics simulations
- PMID: 21500218
- PMCID: PMC3144279
- DOI: 10.1002/jcc.21787
MDAnalysis: a toolkit for the analysis of molecular dynamics simulations
Abstract
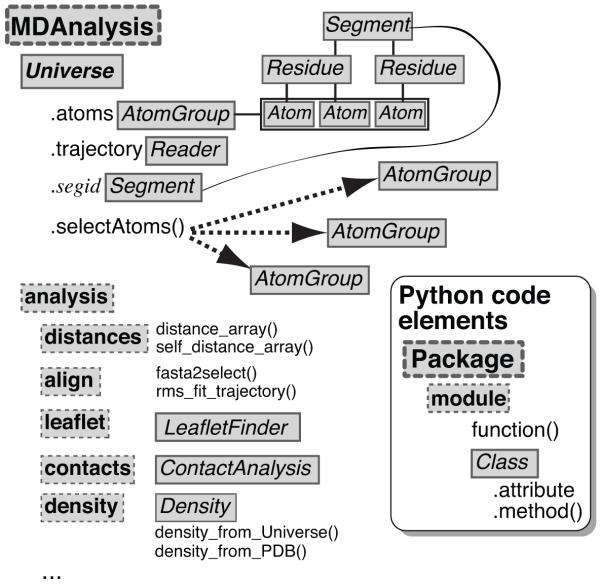
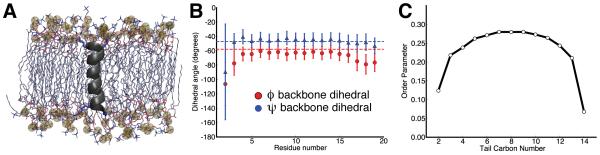
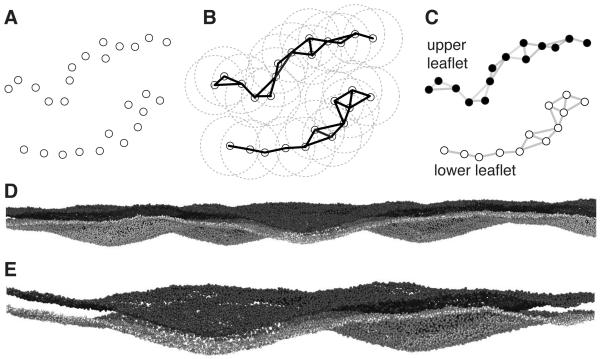
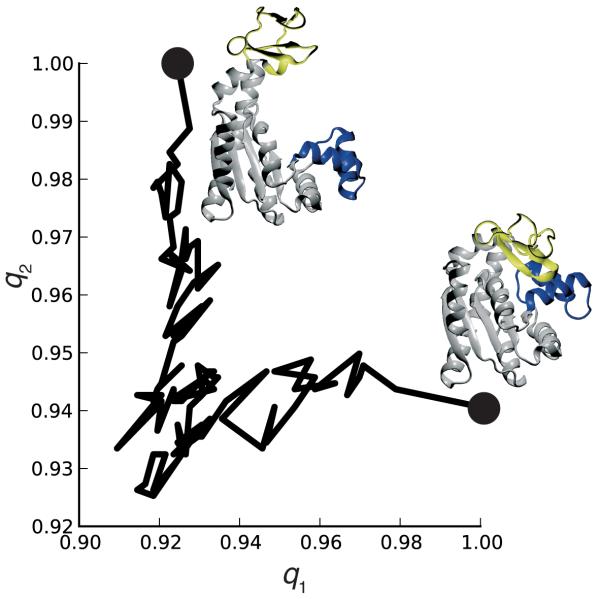
MDAnalysis is an object-oriented library for structural and temporal analysis of molecular dynamics (MD) simulation trajectories and individual protein structures. It is written in the Python language with some performance-critical code in C. It uses the powerful NumPy package to expose trajectory data as fast and efficient NumPy arrays. It has been tested on systems of millions of particles. Many common file formats of simulation packages including CHARMM, Gromacs, Amber, and NAMD and the Protein Data Bank format can be read and written. Atoms can be selected with a syntax similar to CHARMM's powerful selection commands. MDAnalysis enables both novice and experienced programmers to rapidly write their own analytical tools and access data stored in trajectories in an easily accessible manner that facilitates interactive explorative analysis. MDAnalysis has been tested on and works for most Unix-based platforms such as Linux and Mac OS X. It is freely available under the GNU General Public License from http://mdanalysis.googlecode.com.
Keywords: Python programming language; analysis; membrane systems; molecular dynamics simulations; object-oriented design; proteins; software.
Copyright © 2011 Wiley Periodicals, Inc.
Figures

Similar articles
-
LOOS: an extensible platform for the structural analysis of simulations.Annu Int Conf IEEE Eng Med Biol Soc. 2009;2009:2332-5. doi: 10.1109/IEMBS.2009.5335065. Annu Int Conf IEEE Eng Med Biol Soc. 2009. PMID: 19965179
-
Morphoscanner2.0: A new python module for analysis of molecular dynamics simulations.PLoS One. 2023 Apr 27;18(4):e0284307. doi: 10.1371/journal.pone.0284307. eCollection 2023. PLoS One. 2023. PMID: 37104393 Free PMC article.
-
Pytim: A python package for the interfacial analysis of molecular simulations.J Comput Chem. 2018 Sep 30;39(25):2118-2125. doi: 10.1002/jcc.25384. Epub 2018 Oct 10. J Comput Chem. 2018. PMID: 30306571 Free PMC article.
-
Analysis Libraries for Molecular Trajectories: A Cross-Language Synopsis.Methods Mol Biol. 2019;2022:503-527. doi: 10.1007/978-1-4939-9608-7_20. Methods Mol Biol. 2019. PMID: 31396916 Review.
-
Array programming with NumPy.Nature. 2020 Sep;585(7825):357-362. doi: 10.1038/s41586-020-2649-2. Epub 2020 Sep 16. Nature. 2020. PMID: 32939066 Free PMC article. Review.
Cited by
-
Dissected antiporter modules establish minimal proton-conduction elements of the respiratory complex I.Nat Commun. 2024 Oct 22;15(1):9098. doi: 10.1038/s41467-024-53194-5. Nat Commun. 2024. PMID: 39438463 Free PMC article.
-
Membrane association of the PTEN tumor suppressor: electrostatic interaction with phosphatidylserine-containing bilayers and regulatory role of the C-terminal tail.J Struct Biol. 2012 Dec;180(3):394-408. doi: 10.1016/j.jsb.2012.10.003. Epub 2012 Oct 13. J Struct Biol. 2012. PMID: 23073177 Free PMC article.
-
Detecting the First Hydration Shell Structure around Biomolecules at Interfaces.ACS Cent Sci. 2022 Oct 26;8(10):1404-1414. doi: 10.1021/acscentsci.2c00702. Epub 2022 Sep 6. ACS Cent Sci. 2022. PMID: 36313165 Free PMC article.
-
Lipid-mediated activation of plasma membrane-localized deubiquitylating enzymes modulate endosomal trafficking.Nat Commun. 2022 Nov 12;13(1):6897. doi: 10.1038/s41467-022-34637-3. Nat Commun. 2022. PMID: 36371501 Free PMC article.
-
PyMM: An Open-Source Python Program for QM/MM Simulations Based on the Perturbed Matrix Method.J Chem Theory Comput. 2023 Jan 10;19(1):33-41. doi: 10.1021/acs.jctc.2c00767. Epub 2022 Nov 15. J Chem Theory Comput. 2023. PMID: 36378163 Free PMC article.
References
-
- Hess B, Kutzner C, van der Spoel D, Lindahl E. J Chem Theo Comp. 2008;4(3):435–447. - PubMed
-
- Seeber M, Cecchini M, Rao F, Settanni G, Caflisch A. Bioinformatics. 2007;23(19):2625–2627. - PubMed
-
- Verstraelen T, Van Houteghem M, Van Speybroeck V, Waroquier M. J Chem Inform Model. 2008;48(12):2414–2424. - PubMed
-
- Mezei M. J Comput Chem. 2010;31(14):2658–2668. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
