

A comprehensive genomic binding map of gene and chromatin regulatory proteins in Saccharomyces
- PMID: 21329885
- PMCID: PMC3057419
- DOI: 10.1016/j.molcel.2011.01.015
A comprehensive genomic binding map of gene and chromatin regulatory proteins in Saccharomyces
Abstract
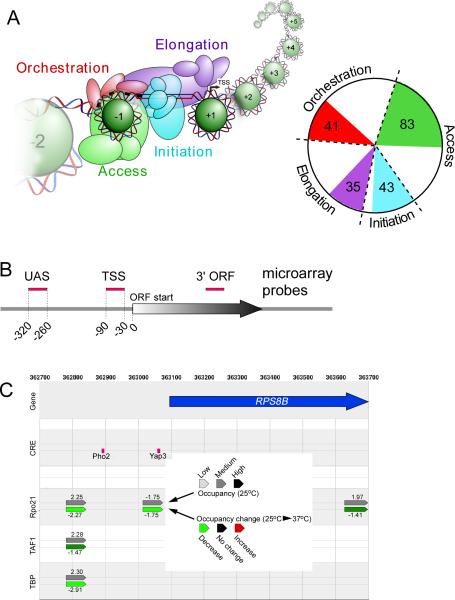
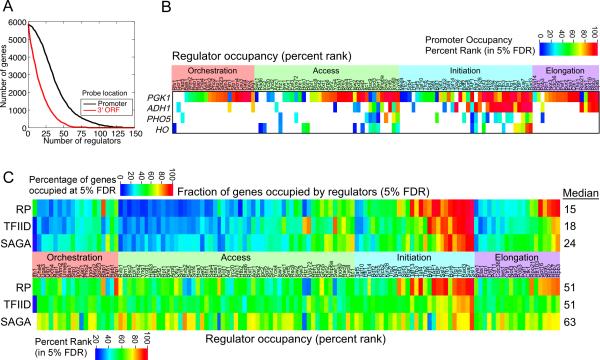
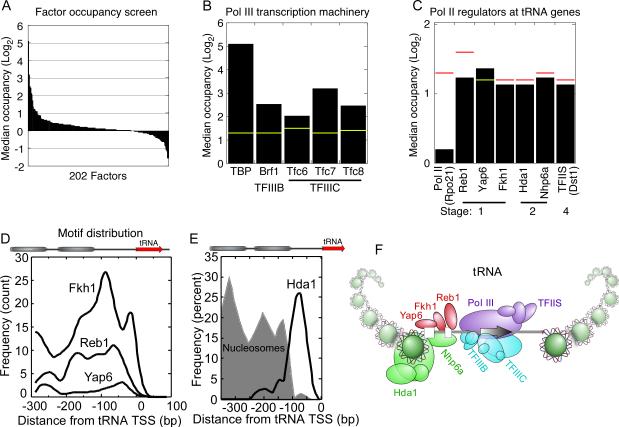
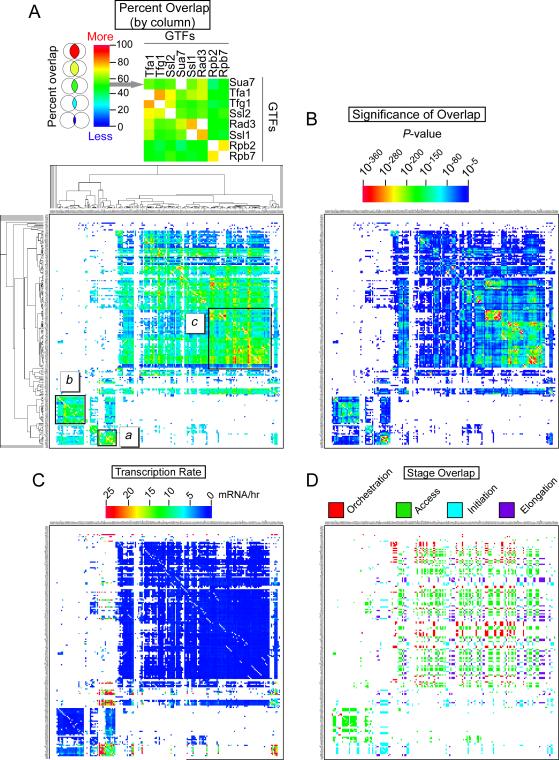
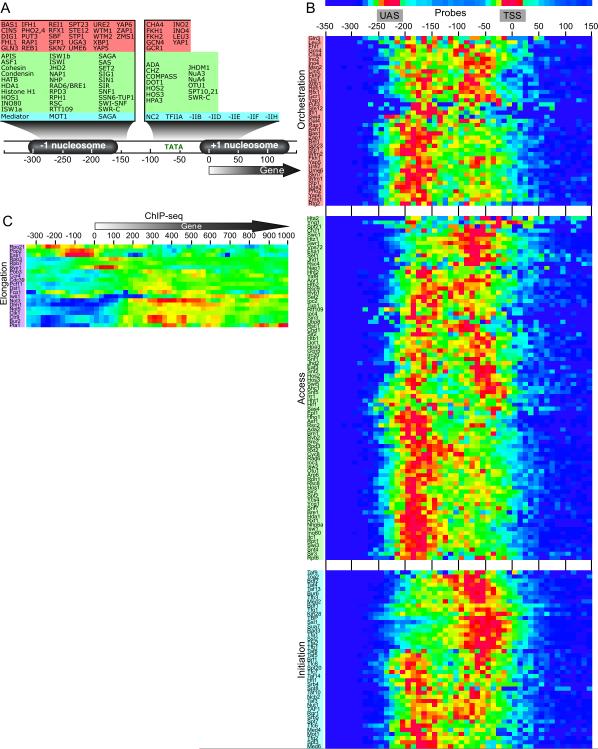
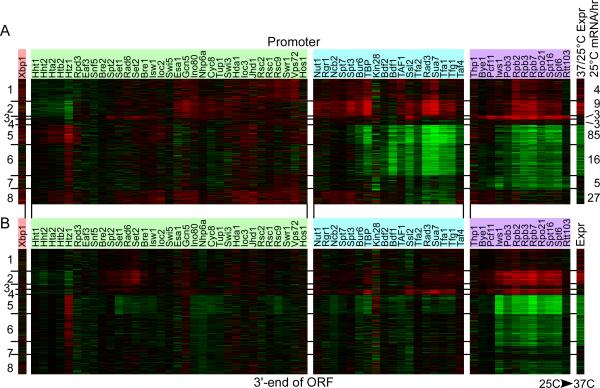
Hundreds of different proteins regulate and implement transcription in Saccharomyces. Yet their interrelationships have not been investigated on a comprehensive scale. Here we determined the genome-wide binding locations of 200 transcription-related proteins, under normal and acute heat-shock conditions. This study distinguishes binding between distal versus proximal promoter regions as well as the 3' ends of genes for nearly all mRNA and tRNA genes. This study reveals (1) a greater diversity and specialization of regulation associated with the SAGA transcription pathway compared to the TFIID pathway, (2) new regulators enriched at tRNA genes, (3) a global co-occupancy network of >20,000 unique regulator combinations that show a high degree of regulatory interconnections among lowly expressed genes, (4) regulators of the SAGA pathway located largely distal to the core promoter and regulators of the TFIID pathway located proximally, and (5) distinct mobilization of SAGA- versus TFIID-linked regulators during acute heat shock.
Copyright © 2011 Elsevier Inc. All rights reserved.
Figures
Similar articles
-
A genome-wide housekeeping role for TFIID and a highly regulated stress-related role for SAGA in Saccharomyces cerevisiae.Mol Cell. 2004 Feb 27;13(4):573-85. doi: 10.1016/s1097-2765(04)00087-5. Mol Cell. 2004. PMID: 14992726
-
Sequential recruitment of SAGA and TFIID in a genomic response to DNA damage in Saccharomyces cerevisiae.Mol Cell Biol. 2011 Jan;31(1):190-202. doi: 10.1128/MCB.00317-10. Epub 2010 Oct 18. Mol Cell Biol. 2011. PMID: 20956559 Free PMC article.
-
Transcription of Nearly All Yeast RNA Polymerase II-Transcribed Genes Is Dependent on Transcription Factor TFIID.Mol Cell. 2017 Oct 5;68(1):118-129.e5. doi: 10.1016/j.molcel.2017.08.014. Epub 2017 Sep 14. Mol Cell. 2017. PMID: 28918900 Free PMC article.
-
SAGA and TFIID: Friends of TBP drifting apart.Biochim Biophys Acta Gene Regul Mech. 2021 Feb;1864(2):194604. doi: 10.1016/j.bbagrm.2020.194604. Epub 2020 Jul 14. Biochim Biophys Acta Gene Regul Mech. 2021. PMID: 32673655 Review.
-
The role of chromatin structure in regulating stress-induced transcription in Saccharomyces cerevisiae.Biochem Cell Biol. 2006 Aug;84(4):477-89. doi: 10.1139/o06-079. Biochem Cell Biol. 2006. PMID: 16936821 Review.
Cited by
-
Painting by numbers: increasing the parts list for chromatin domains.Mol Cell. 2013 Feb 21;49(4):620-1. doi: 10.1016/j.molcel.2013.02.007. Mol Cell. 2013. PMID: 23438859 Free PMC article.
-
Rph1/KDM4 mediates nutrient-limitation signaling that leads to the transcriptional induction of autophagy.Curr Biol. 2015 Mar 2;25(5):546-55. doi: 10.1016/j.cub.2014.12.049. Epub 2015 Feb 5. Curr Biol. 2015. PMID: 25660547 Free PMC article.
-
Kcs1 and Vip1: The Key Enzymes behind Inositol Pyrophosphate Signaling in Saccharomyces cerevisiae.Biomolecules. 2024 Jan 26;14(2):152. doi: 10.3390/biom14020152. Biomolecules. 2024. PMID: 38397389 Free PMC article. Review.
-
Motif discovery and transcription factor binding sites before and after the next-generation sequencing era.Brief Bioinform. 2013 Mar;14(2):225-37. doi: 10.1093/bib/bbs016. Epub 2012 Apr 19. Brief Bioinform. 2013. PMID: 22517426 Free PMC article.
-
YHMI: a web tool to identify histone modifications and histone/chromatin regulators from a gene list in yeast.Database (Oxford). 2018 Jan 1;2018:bay116. doi: 10.1093/database/bay116. Database (Oxford). 2018. PMID: 30371756 Free PMC article.
References
-
- Albert I, Mavrich TN, Tomsho LP, Qi J, Zanton SJ, Schuster SC, Pugh BF. Translational and rotational settings of H2A.Z nucleosomes across the Saccharomyces cerevisiae genome. Nature. 2007;446:572–576. - PubMed
-
- Basehoar AD, Zanton SJ, Pugh BF. Identification and distinct regulation of yeast TATA box-containing genes. Cell. 2004;116:699–709. - PubMed
-
- Berger SL. Gene regulation. Local or global? Nature. 2000;408:412–413. 415. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
