

MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase
- PMID: 21262914
- PMCID: PMC3059327
- DOI: 10.1158/0008-5472.CAN-10-3367
MYC phosphorylation, activation, and tumorigenic potential in hepatocellular carcinoma are regulated by HMG-CoA reductase
Abstract
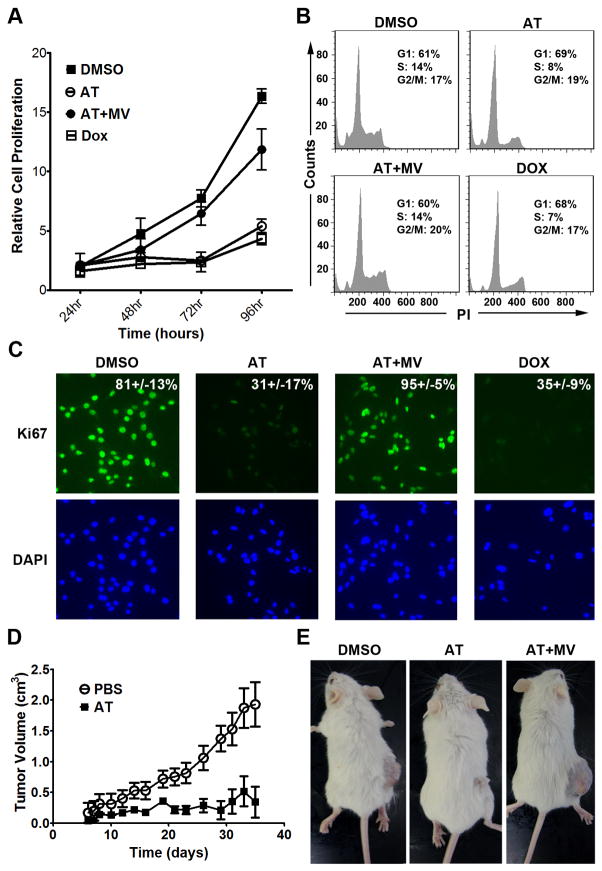
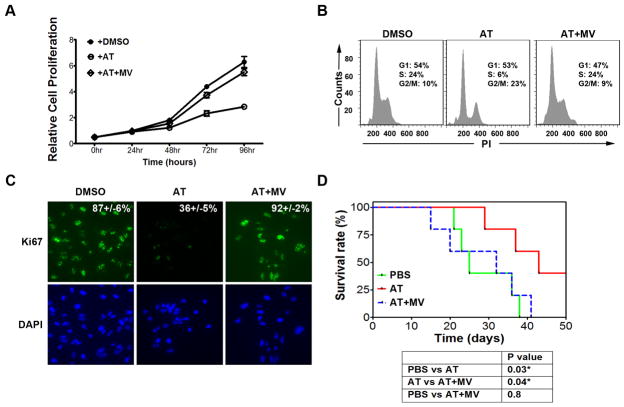
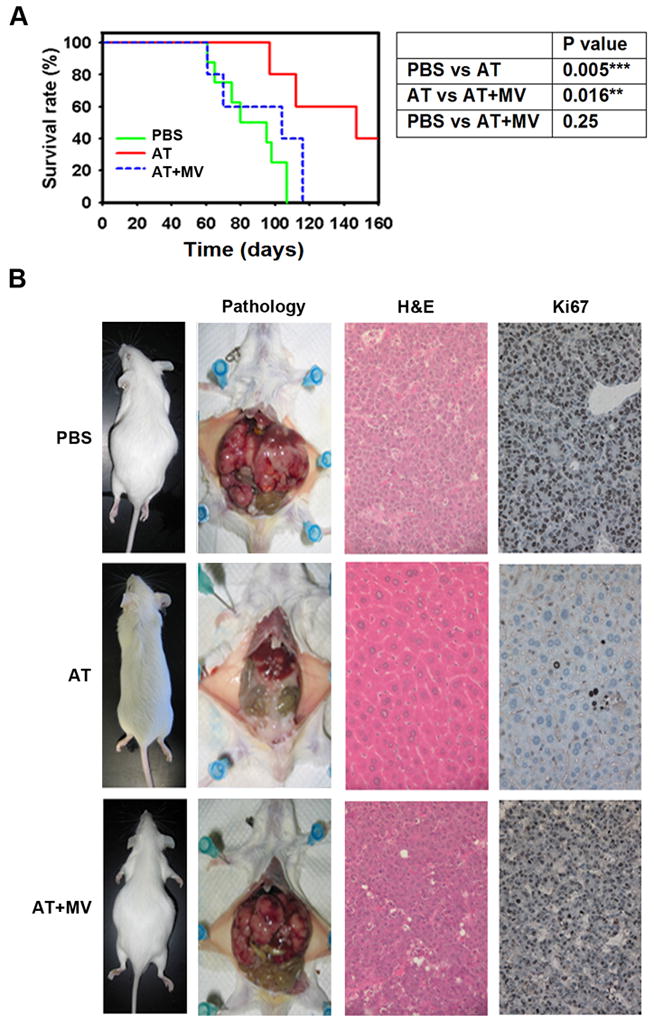
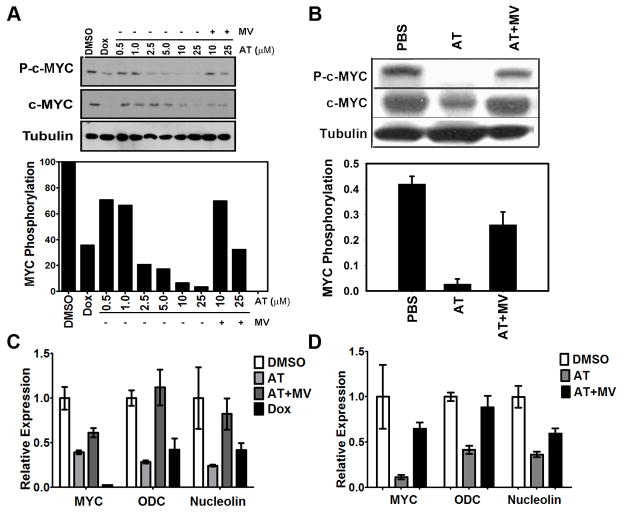
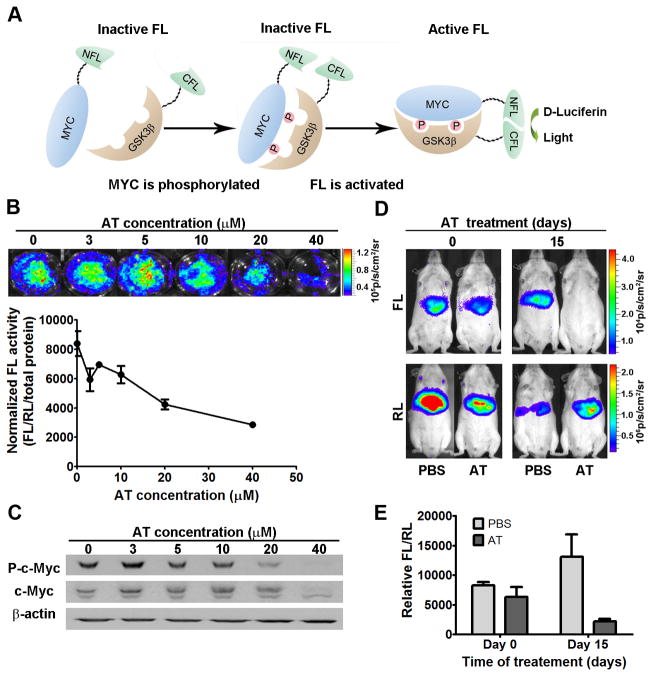
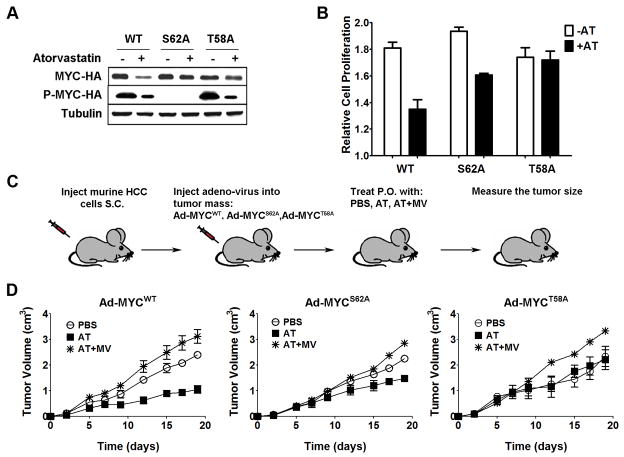
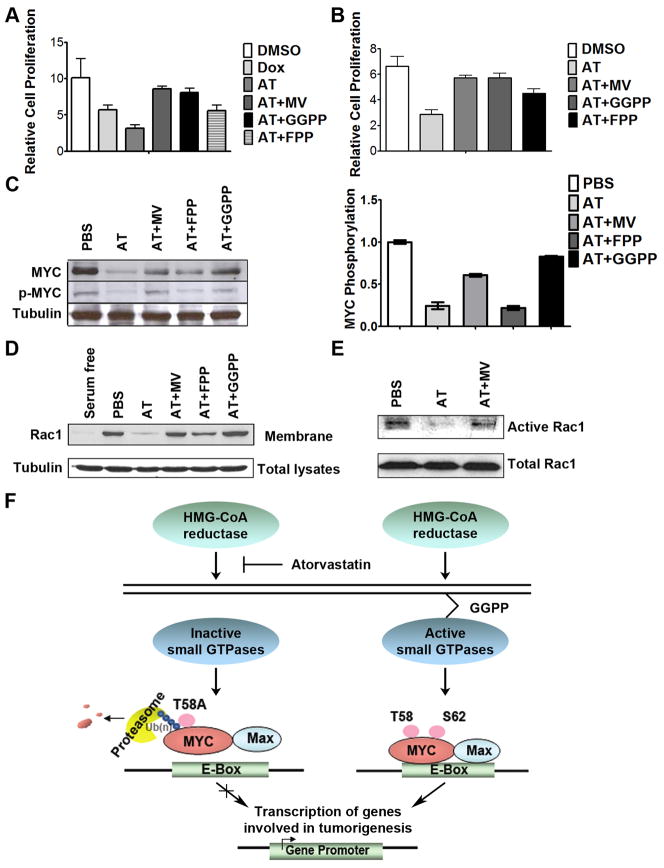
MYC is a potential target for many cancers but is not amenable to existing pharmacologic approaches. Inhibition of 3-hydroxy-3-methylglutaryl-coenzyme A reductase (HMG-CoA reductase) by statins has shown potential efficacy against a number of cancers. Here, we show that inhibition of HMG-CoA reductase by atorvastatin (AT) blocks both MYC phosphorylation and activation, suppressing tumor initiation and growth in vivo in a transgenic model of MYC-induced hepatocellular carcinoma (HCC) as well as in human HCC-derived cell lines. To confirm specificity, we show that the antitumor effects of AT are blocked by cotreatment with the HMG-CoA reductase product mevalonate. Moreover, by using a novel molecular imaging sensor, we confirm that inhibition of HMG-CoA reductase blocks MYC phosphorylation in vivo. Importantly, the introduction of phosphorylation mutants of MYC at Ser62 or Thr58 into tumors blocks their sensitivity to inhibition of HMG-CoA reductase. Finally, we show that inhibition of HMG-CoA reductase suppresses MYC phosphorylation through Rac GTPase. Therefore, HMG-CoA reductase is a critical regulator of MYC phosphorylation, activation, and tumorigenic properties. The inhibition of HMG-CoA reductase may be a useful target for the treatment of MYC-associated HCC as well as other tumors.
© 2011 AACR.
Conflict of interest statement
Figures
Similar articles
-
Inhibition of HMGcoA reductase by atorvastatin prevents and reverses MYC-induced lymphomagenesis.Blood. 2007 Oct 1;110(7):2674-84. doi: 10.1182/blood-2006-09-048033. Epub 2007 Jul 10. Blood. 2007. PMID: 17622571 Free PMC article.
-
Inhibition of MEK suppresses hepatocellular carcinoma growth through independent MYC and BIM regulation.Cell Oncol (Dordr). 2019 Jun;42(3):369-380. doi: 10.1007/s13402-019-00432-4. Epub 2019 Feb 20. Cell Oncol (Dordr). 2019. PMID: 30788663
-
Automated enzyme inhibition assay method for the determination of atorvastatin-derived HMG-CoA reductase inhibitors in human plasma using radioactivity detection.J Pharmacol Toxicol Methods. 2008 Jan-Feb;57(1):61-9. doi: 10.1016/j.vascn.2007.06.002. Epub 2007 Jun 23. J Pharmacol Toxicol Methods. 2008. PMID: 17651990
-
Atorvastatin: pharmacological characteristics and lipid-lowering effects.Drugs. 2007;67 Suppl 1:3-15. doi: 10.2165/00003495-200767001-00002. Drugs. 2007. PMID: 17910517 Review.
-
Repositioning of HMG-CoA Reductase Inhibitors as Adjuvants in the Modulation of Efflux Pump-Mediated Bacterial and Tumor Resistance.Antibiotics (Basel). 2023 Sep 20;12(9):1468. doi: 10.3390/antibiotics12091468. Antibiotics (Basel). 2023. PMID: 37760764 Free PMC article. Review.
Cited by
-
Dual Targeting of 3-Hydroxy-3-methylglutaryl Coenzyme A Reductase and Histone Deacetylase as a Therapy for Colorectal Cancer.EBioMedicine. 2016 Aug;10:124-36. doi: 10.1016/j.ebiom.2016.07.019. Epub 2016 Jul 17. EBioMedicine. 2016. PMID: 27448759 Free PMC article.
-
The Impact of Statin Use and Breast Cancer Recurrence - A Retrospective Study in Singapore.Front Oncol. 2022 Mar 31;12:835320. doi: 10.3389/fonc.2022.835320. eCollection 2022. Front Oncol. 2022. PMID: 35433431 Free PMC article.
-
Effect of Statins on Lung Cancer Molecular Pathways: A Possible Therapeutic Role.Pharmaceuticals (Basel). 2022 May 10;15(5):589. doi: 10.3390/ph15050589. Pharmaceuticals (Basel). 2022. PMID: 35631415 Free PMC article. Review.
-
Cholesterol biosynthesis supports the growth of hepatocarcinoma lesions depleted of fatty acid synthase in mice and humans.Gut. 2020 Jan;69(1):177-186. doi: 10.1136/gutjnl-2018-317581. Epub 2019 Apr 6. Gut. 2020. PMID: 30954949 Free PMC article.
-
Hypoxia Pathway Proteins As Central Mediators of Metabolism in the Tumor Cells and Their Microenvironment.Front Immunol. 2018 Jan 29;9:40. doi: 10.3389/fimmu.2018.00040. eCollection 2018. Front Immunol. 2018. PMID: 29434587 Free PMC article. Review.
References
-
- Farazi PA, DePinho RA. Hepatocellular carcinoma pathogenesis: from genes to environment. Nat Rev Cancer. 2006;6:674–87. - PubMed
-
- Fausto N, Campbell JS, Riehle KJ. Liver regeneration. Hepatology. 2006;43:S45–53. - PubMed
-
- Coleman WB. Mechanisms of human hepatocarcinogenesis. Curr Mol Med. 2003;3:573–88. - PubMed
-
- Lee JS, Chu IS, Mikaelyan A, et al. Application of comparative functional genomics to identify best-fit mouse models to study human cancer. Nat Genet. 2004;36:1306–11. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- CA114747/CA/NCI NIH HHS/United States
- U56 CA112973/CA/NCI NIH HHS/United States
- CA89305-01A1/CA/NCI NIH HHS/United States
- F32-CA132312/CA/NCI NIH HHS/United States
- CA112973/CA/NCI NIH HHS/United States
- R01 CA089305/CA/NCI NIH HHS/United States
- F32 CA132312/CA/NCI NIH HHS/United States
- R01 CA105102/CA/NCI NIH HHS/United States
- CA105102/CA/NCI NIH HHS/United States
- CA89305-0351/CA/NCI NIH HHS/United States
- R01 CA089305-09/CA/NCI NIH HHS/United States
- R01 CA105102-06/CA/NCI NIH HHS/United States
- T32 AI007290/AI/NIAID NIH HHS/United States
- R01 CA105102-04/CA/NCI NIH HHS/United States
- P50 CA114747/CA/NCI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Miscellaneous
