

PPAR-γ ligands repress TGFβ-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis
- PMID: 21253589
- PMCID: PMC3017065
- DOI: 10.1371/journal.pone.0015909
PPAR-γ ligands repress TGFβ-induced myofibroblast differentiation by targeting the PI3K/Akt pathway: implications for therapy of fibrosis
Abstract
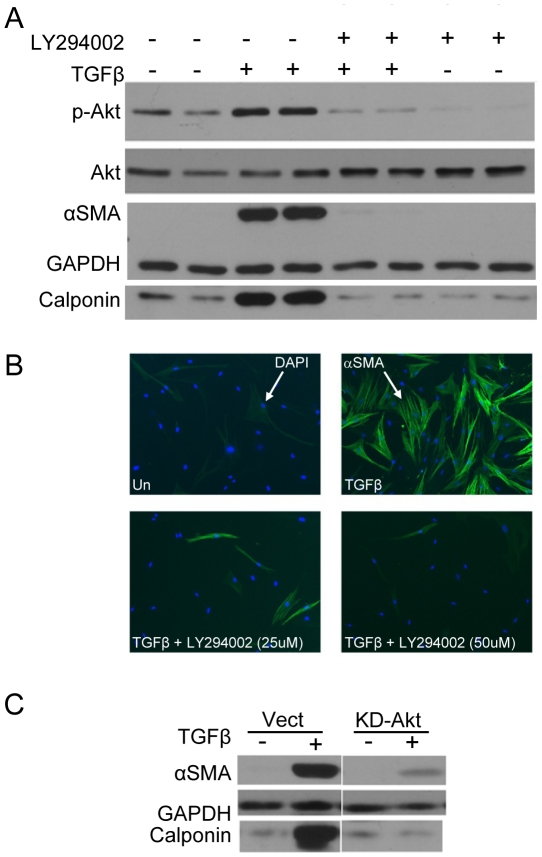
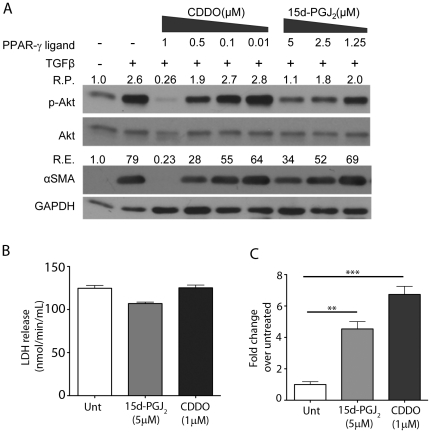
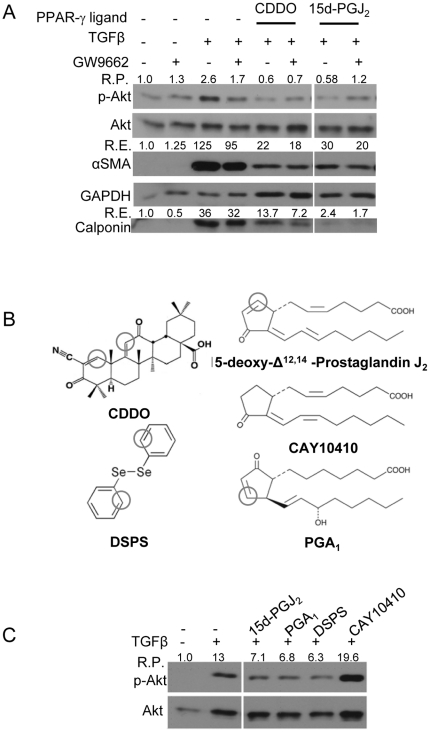
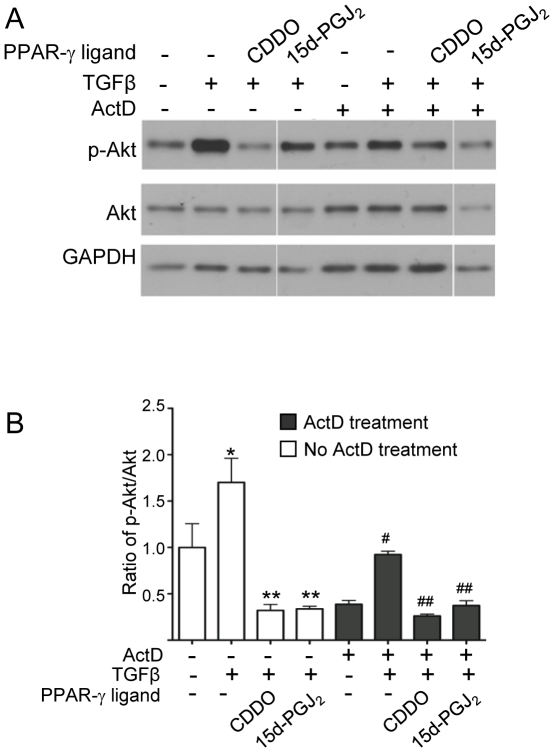
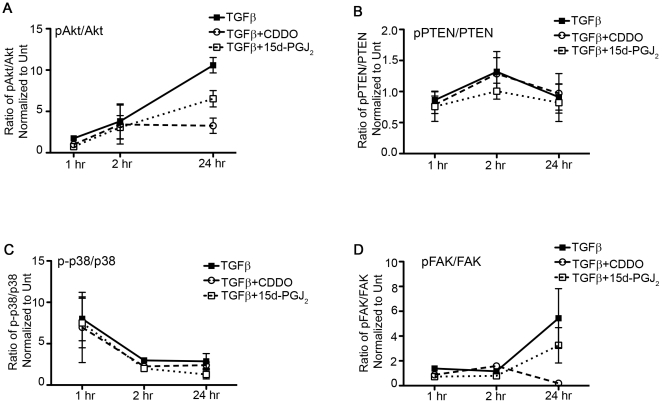
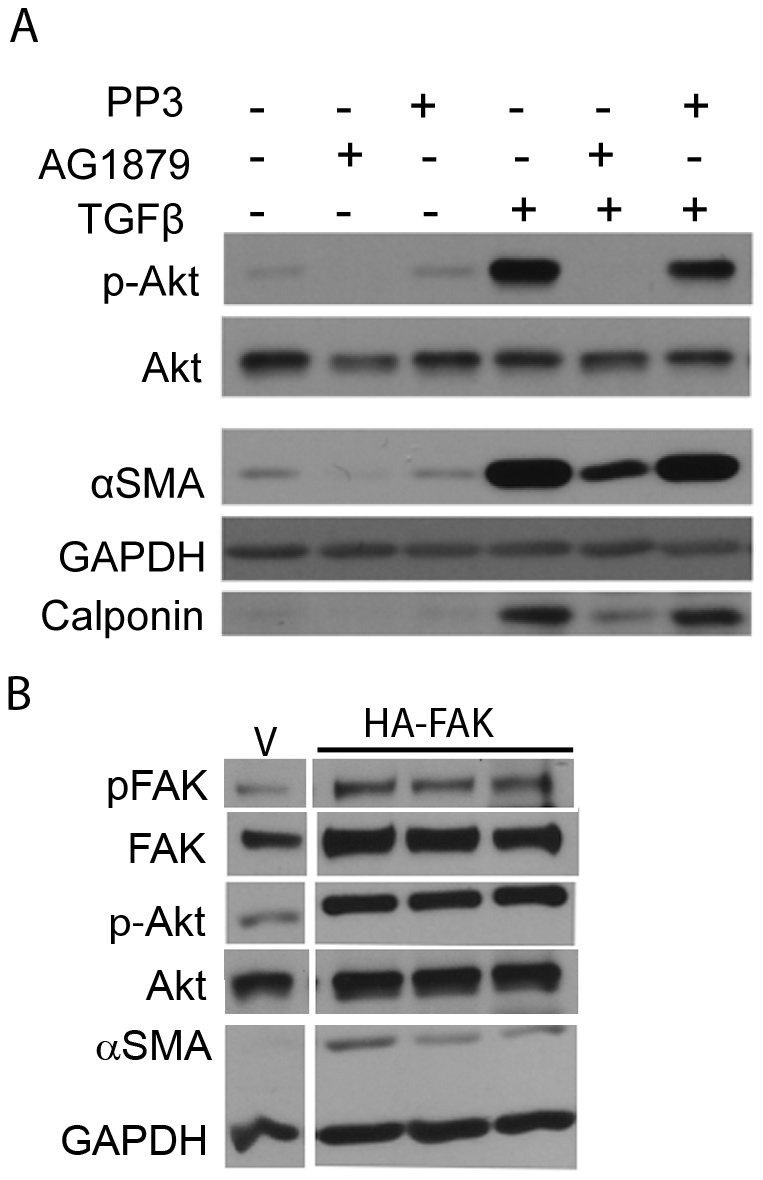
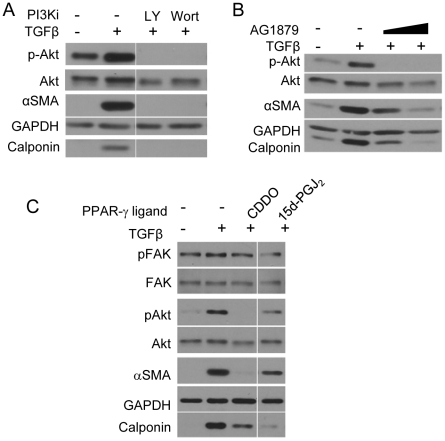
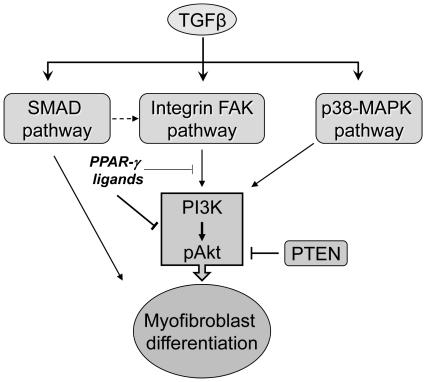
Transforming growth factor beta (TGFβ) induced differentiation of human lung fibroblasts to myofibroblasts is a key event in the pathogenesis of pulmonary fibrosis. Although the typical TGFβ signaling pathway involves the Smad family of transcription factors, we have previously reported that peroxisome proliferator-activated receptor-γ (PPAR-γ) ligands inhibit TGFβ-mediated differentiation of human lung fibroblasts to myofibroblasts via a Smad-independent pathway. TGFβ also activates the phosphatidylinositol 3 kinase/protein kinase B (PI3K/Akt) pathway leading to phosphorylation of Akt(S473). Here, we report that PPAR-γ ligands, 2-cyano-3,12-dioxooleana-1,9-dien-28-oic acid (CDDO) and 15-deoxy-(12,14)-15d-prostaglandin J(2) (15d-PGJ(2)), inhibit human myofibroblast differentiation of normal and idiopathic pulmonary fibrotic (IPF) fibroblasts, by blocking Akt phosphorylation at Ser473 by a PPAR-γ-independent mechanism. The PI3K inhibitor LY294002 and a dominant-negative inactive kinase-domain mutant of Akt both inhibited TGFβ-stimulated myofibroblast differentiation, as determined by Western blotting for α-smooth muscle actin and calponin. Prostaglandin A(1) (PGA(1)), a structural analogue of 15d-PGJ(2) with an electrophilic center, also reduced TGFβ-driven phosphorylation of Akt, while CAY10410, another analogue that lacks an electrophilic center, did not; implying that the activity of 15d-PGJ(2) and CDDO is dependent on their electrophilic properties. PPAR-γ ligands inhibited TGFβ-induced Akt phosphorylation via both post-translational and post-transcriptional mechanisms. This inhibition is independent of MAPK-p38 and PTEN but is dependent on TGFβ-induced phosphorylation of FAK, a kinase that acts upstream of Akt. Thus, PPAR-γ ligands inhibit TGFβ signaling by affecting two pro-survival pathways that culminate in myofibroblast differentiation. Further studies of PPAR-γ ligands and small electrophilic molecules may lead to a new generation of anti-fibrotic therapeutics.
Conflict of interest statement
Figures
Similar articles
-
Electrophilic peroxisome proliferator-activated receptor-gamma ligands have potent antifibrotic effects in human lung fibroblasts.Am J Respir Cell Mol Biol. 2009 Dec;41(6):722-30. doi: 10.1165/rcmb.2009-0006OC. Epub 2009 Mar 13. Am J Respir Cell Mol Biol. 2009. PMID: 19286977 Free PMC article.
-
Electrophilic PPARγ ligands inhibit corneal fibroblast to myofibroblast differentiation in vitro: a potentially novel therapy for corneal scarring.Exp Eye Res. 2012 Jan;94(1):136-45. doi: 10.1016/j.exer.2011.11.018. Epub 2011 Dec 8. Exp Eye Res. 2012. PMID: 22178289 Free PMC article.
-
A synthetic PPAR-γ agonist triterpenoid ameliorates experimental fibrosis: PPAR-γ-independent suppression of fibrotic responses.Ann Rheum Dis. 2014 Feb;73(2):446-54. doi: 10.1136/annrheumdis-2012-202716. Epub 2013 Mar 20. Ann Rheum Dis. 2014. PMID: 23515440 Free PMC article.
-
Interactions between TGF-β1, canonical WNT/β-catenin pathway and PPAR γ in radiation-induced fibrosis.Oncotarget. 2017 Sep 23;8(52):90579-90604. doi: 10.18632/oncotarget.21234. eCollection 2017 Oct 27. Oncotarget. 2017. PMID: 29163854 Free PMC article. Review.
-
PPARγ and TGFβ-Major Regulators of Metabolism, Inflammation, and Fibrosis in the Lungs and Kidneys.Int J Mol Sci. 2021 Sep 28;22(19):10431. doi: 10.3390/ijms221910431. Int J Mol Sci. 2021. PMID: 34638771 Free PMC article. Review.
Cited by
-
The Role of Nrf2 in Pulmonary Fibrosis: Molecular Mechanisms and Treatment Approaches.Antioxidants (Basel). 2022 Aug 29;11(9):1685. doi: 10.3390/antiox11091685. Antioxidants (Basel). 2022. PMID: 36139759 Free PMC article. Review.
-
Necrotizing Enterocolitis: LPS/TLR4-Induced Crosstalk Between Canonical TGF-β/Wnt/β-Catenin Pathways and PPARγ.Front Pediatr. 2021 Oct 12;9:713344. doi: 10.3389/fped.2021.713344. eCollection 2021. Front Pediatr. 2021. PMID: 34712628 Free PMC article. Review.
-
Bronchopulmonary Dysplasia: Crosstalk Between PPARγ, WNT/β-Catenin and TGF-β Pathways; The Potential Therapeutic Role of PPARγ Agonists.Front Pediatr. 2019 May 3;7:176. doi: 10.3389/fped.2019.00176. eCollection 2019. Front Pediatr. 2019. PMID: 31131268 Free PMC article. Review.
-
Selenium and Selenoproteins in Gut Inflammation-A Review.Antioxidants (Basel). 2018 Mar 1;7(3):36. doi: 10.3390/antiox7030036. Antioxidants (Basel). 2018. PMID: 29494512 Free PMC article. Review.
-
The emerging roles of β-arrestins in fibrotic diseases.Acta Pharmacol Sin. 2015 Nov;36(11):1277-87. doi: 10.1038/aps.2015.74. Epub 2015 Sep 21. Acta Pharmacol Sin. 2015. PMID: 26388156 Free PMC article. Review.
References
-
- Sime PJ, O'Reilly KM. Fibrosis of the lung and other tissues: new concepts in pathogenesis and treatment. Clin Immunol. 2001;99:308–319. - PubMed
-
- Baglole CJ, Ray DM, Bernstein SH, Feldon SE, Smith TJ, et al. More than structural cells, fibroblasts create and orchestrate the tumor microenvironment. Immunol Invest. 2006;35:297–325. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
- HL095402-02/HL/NHLBI NIH HHS/United States
- HL075432/HL/NHLBI NIH HHS/United States
- P30 ES001247/ES/NIEHS NIH HHS/United States
- HL075432-04S1/HL/NHLBI NIH HHS/United States
- R03 HL095402/HL/NHLBI NIH HHS/United States
- F30 HL097596/HL/NHLBI NIH HHS/United States
- T32 HL66988/HL/NHLBI NIH HHS/United States
- R01 HL075432/HL/NHLBI NIH HHS/United States
- P30 ES01247/ES/NIEHS NIH HHS/United States
- T32 ES007026/ES/NIEHS NIH HHS/United States
- T32 HL066988/HL/NHLBI NIH HHS/United States
- T32 ES07026/ES/NIEHS NIH HHS/United States
- HL075432-04S2/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
