

Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy
- PMID: 21252228
- PMCID: PMC3060532
- DOI: 10.1074/jbc.M110.132514
Mutant A53T alpha-synuclein induces neuronal death by increasing mitochondrial autophagy
Abstract
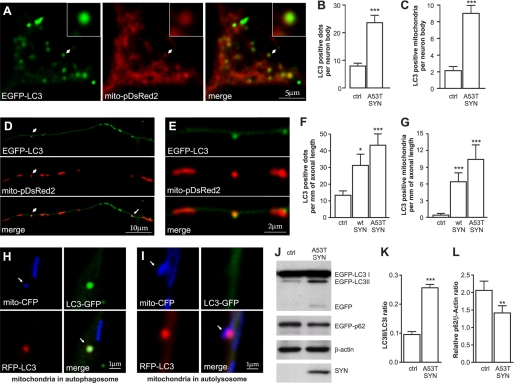
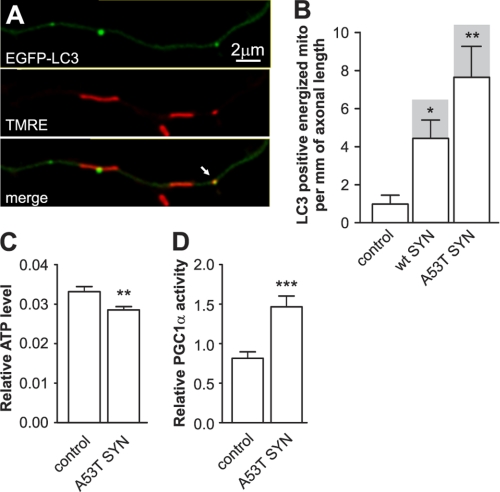
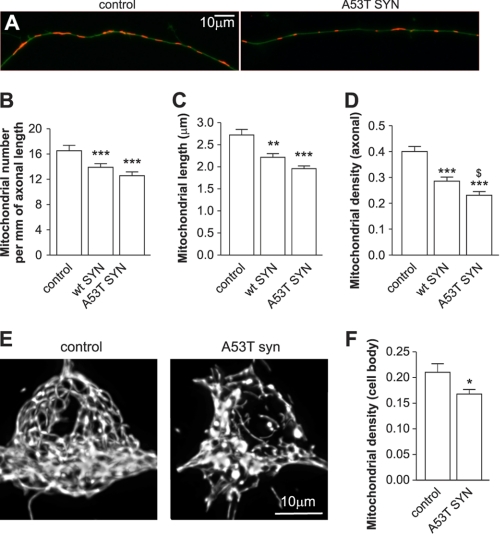
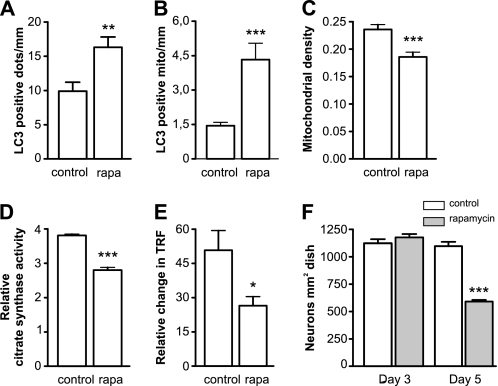
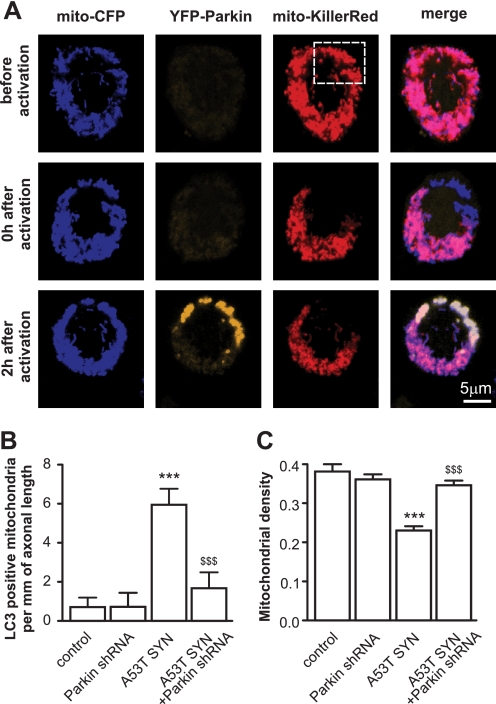
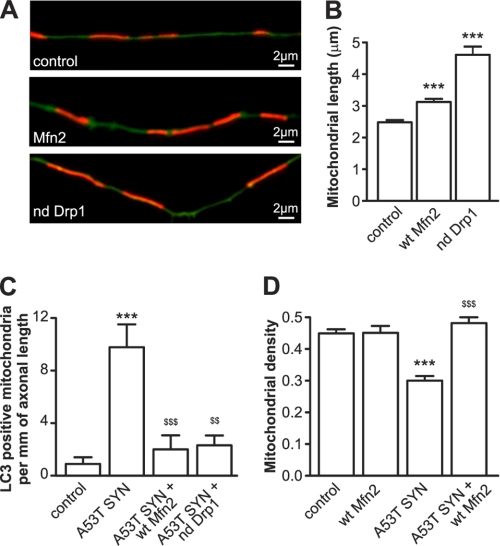
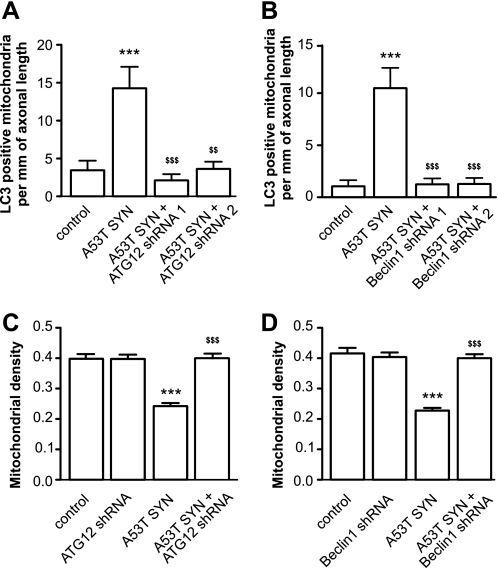
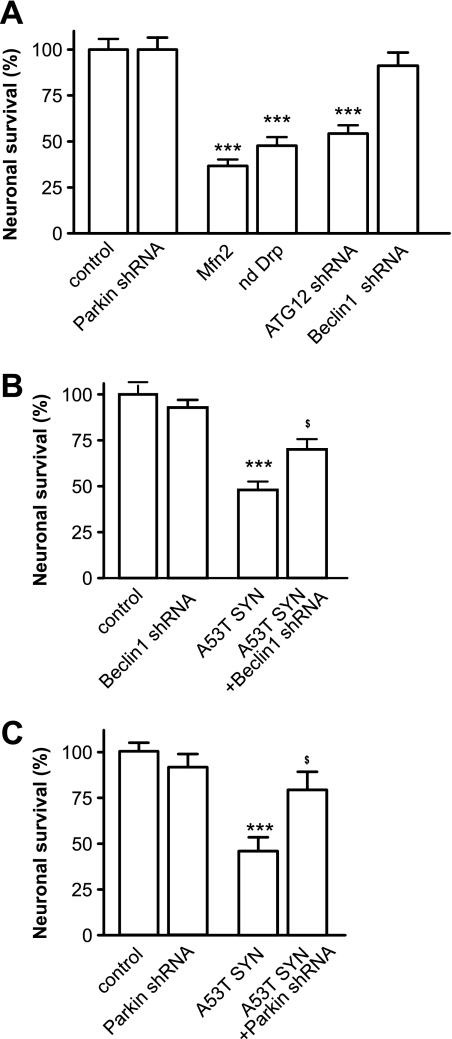
Parkinson disease is characterized by the accumulation of aggregated α-synuclein as the major component of the Lewy bodies. α-Synuclein accumulation in turn leads to compensatory effects that may include the up-regulation of autophagy. Another common feature of Parkinson disease (PD) is mitochondrial dysfunction. Here, we provide evidence that the overactivation of autophagy may be a link that connects the intracellular accumulation of α-synuclein with mitochondrial dysfunction. We found that the activation of macroautophagy in primary cortical neurons that overexpress mutant A53T α-synuclein leads to massive mitochondrial destruction and loss, which is associated with a bioenergetic deficit and neuronal degeneration. No mitochondrial removal or net loss was observed when we suppressed the targeting of mitochondria to autophagosomes by silencing Parkin, overexpressing wild-type Mitofusin 2 and dominant negative Dynamin-related protein 1 or blocking autophagy by silencing autophagy-related genes. The inhibition of targeting mitochondria to autophagosomes or autophagy was also partially protective against mutant A53T α-synuclein-induced neuronal cell death. These data suggest that overactivated mitochondrial removal could be one of the contributing factors that leads to the mitochondrial loss observed in PD models.
Figures
Similar articles
-
Phosphorylation of Parkin at serine 131 by p38 MAPK promotes mitochondrial dysfunction and neuronal death in mutant A53T α-synuclein model of Parkinson's disease.Cell Death Dis. 2018 Jun 13;9(6):700. doi: 10.1038/s41419-018-0722-7. Cell Death Dis. 2018. PMID: 29899409 Free PMC article.
-
Drp-1 dependent mitochondrial fragmentation and protective autophagy in dopaminergic SH-SY5Y cells overexpressing alpha-synuclein.Mol Cell Neurosci. 2018 Apr;88:107-117. doi: 10.1016/j.mcn.2018.01.004. Epub 2018 Feb 3. Mol Cell Neurosci. 2018. PMID: 29414102
-
A53T human α-synuclein overexpression in transgenic mice induces pervasive mitochondria macroautophagy defects preceding dopamine neuron degeneration.J Neurosci. 2015 Jan 21;35(3):890-905. doi: 10.1523/JNEUROSCI.0089-14.2015. J Neurosci. 2015. PMID: 25609609 Free PMC article.
-
Reprint of: revisiting oxidative stress and mitochondrial dysfunction in the pathogenesis of Parkinson disease-resemblance to the effect of amphetamine drugs of abuse.Free Radic Biol Med. 2013 Sep;62:186-201. doi: 10.1016/j.freeradbiomed.2013.05.042. Epub 2013 Jun 3. Free Radic Biol Med. 2013. PMID: 23743292 Review.
-
The Parkinson disease protein α-synuclein inhibits autophagy.Autophagy. 2011 Apr;7(4):429-31. doi: 10.4161/auto.7.4.14393. Epub 2011 Apr 1. Autophagy. 2011. PMID: 21157184 Free PMC article. Review.
Cited by
-
Integrating pathways of Parkinson's disease in a molecular interaction map.Mol Neurobiol. 2014 Feb;49(1):88-102. doi: 10.1007/s12035-013-8489-4. Epub 2013 Jul 7. Mol Neurobiol. 2014. PMID: 23832570 Free PMC article. Review.
-
Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome.PLoS Biol. 2016 Jul 19;14(7):e1002511. doi: 10.1371/journal.pbio.1002511. eCollection 2016 Jul. PLoS Biol. 2016. PMID: 27434582 Free PMC article.
-
Recent advances in bioluminescent probes for neurobiology.Neurophotonics. 2024 Apr;11(2):024204. doi: 10.1117/1.NPh.11.2.024204. Epub 2024 Feb 22. Neurophotonics. 2024. PMID: 38390217 Free PMC article.
-
Bcl-2 homologue Debcl enhances α-synuclein-induced phenotypes in Drosophila.PeerJ. 2016 Sep 15;4:e2461. doi: 10.7717/peerj.2461. eCollection 2016. PeerJ. 2016. PMID: 27672511 Free PMC article.
-
Pre-clinical Studies Identifying Molecular Pathways of Neuroinflammation in Parkinson's Disease: A Systematic Review.Front Aging Neurosci. 2022 Jul 4;14:855776. doi: 10.3389/fnagi.2022.855776. eCollection 2022. Front Aging Neurosci. 2022. PMID: 35912090 Free PMC article.
References
-
- Schapira A. H., Cooper J. M., Dexter D., Jenner P., Clark J. B., Marsden C. D. (1989) Lancet 1, 1269. - PubMed
-
- Henchcliffe C., Beal M. F. (2008) Nat. Clin. Pract. Neurol. 4, 600–609 - PubMed
-
- Ayala A., Venero J. L., Cano J., Machado A. (2007) Front Biosci. 12, 986–1007 - PubMed
-
- Schapira A. H. (2008) Lancet Neurol. 7, 97–109 - PubMed
-
- Anglade P., Vyas S., Javoy-Agid F., Herrero M. T., Michel P. P., Marquez J., Mouatt-Prigent A., Ruberg M., Hirsch E. C., Agid Y. (1997) Histol. Histopathol. 12, 25–31 - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
