

α1AMP-activated protein kinase preserves endothelial function during chronic angiotensin II treatment by limiting Nox2 upregulation
- PMID: 21205985
- PMCID: PMC3066307
- DOI: 10.1161/ATVBAHA.110.219543
α1AMP-activated protein kinase preserves endothelial function during chronic angiotensin II treatment by limiting Nox2 upregulation
Abstract
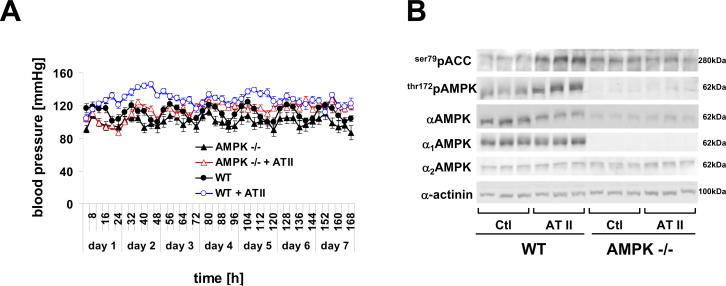
Objective: Besides its well-described metabolic effects, vascular AMP-activated protein kinase (AMPK) can activate endothelial NO synthase, promotes angiogenesis, and limits endothelial cell apoptosis. The current study was designed to study the effects of α1AMPK deletion during vascular disease in vivo.
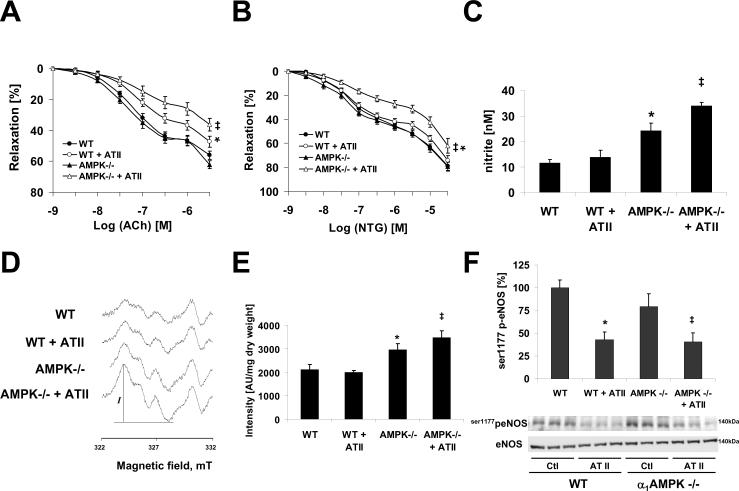
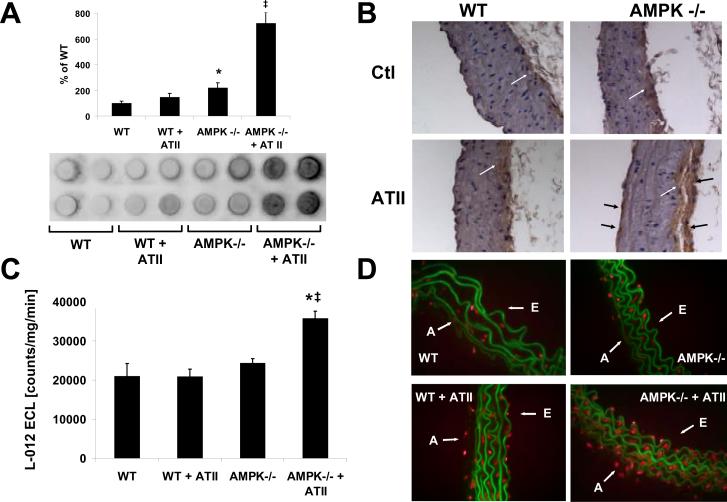
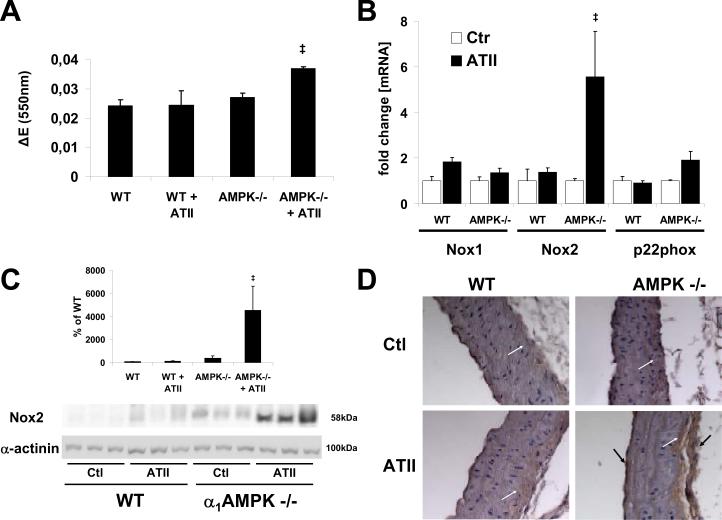
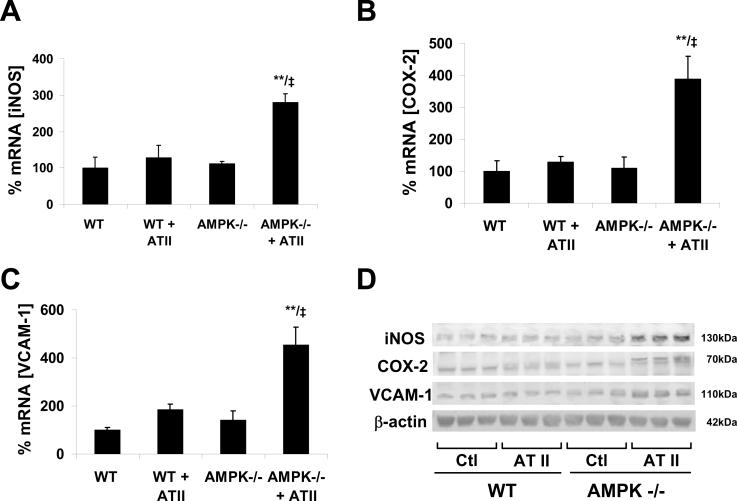
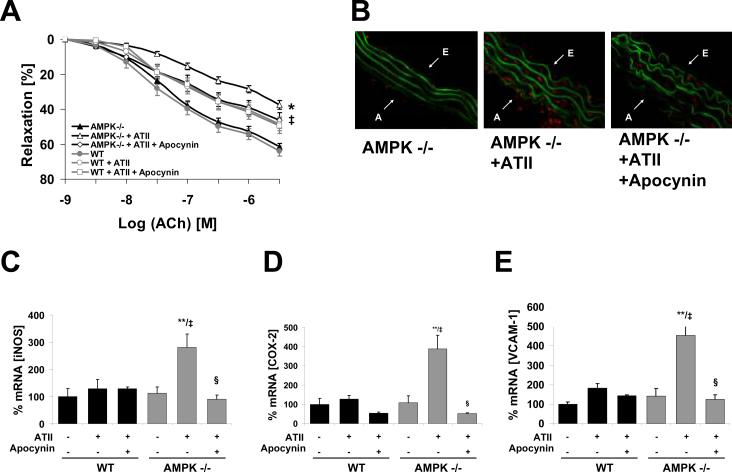
Methods and results: Chronic angiotensin II infusion at low subpressor doses caused a mild endothelial dysfunction that was significantly aggravated in α1AMPK-knockout mice. Unexpectedly, this endothelial dysfunction was not associated with decreased NO content, because NO levels measured by serum nitrite or electron paramagnetic resonance were even increased. However, because of parallel superoxide production, NO was consumed under production of peroxynitrite in angiotensin II-treated α1AMPK-knockout mice, associated with NADPH oxidase activation and Nox2 upregulation. As Nox2 is also a component of phagocyte NADPH oxidases, we found a vascular upregulation of several proinflammatory markers, including inducible NO synthase, vascular cell adhesion molecule-1, and cyclooxygenase-2. Cotreatment with the NADPH oxidase inhibitor apocynin was able to prevent vascular inflammation and also partially restored endothelial function in α1AMPK-knockout mice.
Conclusions: Our data indicate that in vivo α1AMPK deletion leads to Nox2 upregulation, resulting in endothelial dysfunction and vascular inflammation. This implicates basal AMPK activity as a protective, redox-regulating element in vascular homeostasis.
Figures
Similar articles
-
AMPKalpha2 deletion causes aberrant expression and activation of NAD(P)H oxidase and consequent endothelial dysfunction in vivo: role of 26S proteasomes.Circ Res. 2010 Apr 2;106(6):1117-28. doi: 10.1161/CIRCRESAHA.109.212530. Epub 2010 Feb 18. Circ Res. 2010. PMID: 20167927 Free PMC article.
-
Effects of angiotensin II infusion on the expression and function of NAD(P)H oxidase and components of nitric oxide/cGMP signaling.Circ Res. 2002 Mar 8;90(4):E58-65. doi: 10.1161/01.res.0000012569.55432.02. Circ Res. 2002. PMID: 11884382
-
Endothelial Nox2 overexpression potentiates vascular oxidative stress and hemodynamic response to angiotensin II: studies in endothelial-targeted Nox2 transgenic mice.Circ Res. 2007 Apr 13;100(7):1016-25. doi: 10.1161/01.RES.0000263381.83835.7b. Epub 2007 Mar 15. Circ Res. 2007. PMID: 17363703
-
The Nox family of NADPH oxidases: friend or foe of the vascular system?Curr Hypertens Rep. 2012 Feb;14(1):70-8. doi: 10.1007/s11906-011-0238-3. Curr Hypertens Rep. 2012. PMID: 22071588 Review.
-
Oxidative stress, redox signalling and endothelial dysfunction in ageing-related neurodegenerative diseases: a role of NADPH oxidase 2.Br J Clin Pharmacol. 2014 Sep;78(3):441-53. doi: 10.1111/bcp.12357. Br J Clin Pharmacol. 2014. PMID: 25279404 Free PMC article. Review.
Cited by
-
Toll-Like Receptor 4 (TLR4) and AMPK Relevance in Cardiovascular Disease.Adv Pharm Bull. 2023 Jan;13(1):36-47. doi: 10.34172/apb.2023.004. Epub 2021 Oct 10. Adv Pharm Bull. 2023. PMID: 36721803 Free PMC article. Review.
-
Metformin mitigates gas explosion-induced blast lung injuries through AMPK-mediated energy metabolism and NOX2-related oxidation pathway in rats.Exp Ther Med. 2022 Jun 20;24(2):529. doi: 10.3892/etm.2022.11456. eCollection 2022 Aug. Exp Ther Med. 2022. PMID: 35837050 Free PMC article.
-
Nox4 and diabetic nephropathy: with a friend like this, who needs enemies?Free Radic Biol Med. 2013 Aug;61:130-42. doi: 10.1016/j.freeradbiomed.2013.03.014. Epub 2013 Mar 23. Free Radic Biol Med. 2013. PMID: 23528476 Free PMC article. Review.
-
Dual Roles of the AMP-Activated Protein Kinase Pathway in Angiogenesis.Cells. 2019 Jul 19;8(7):752. doi: 10.3390/cells8070752. Cells. 2019. PMID: 31331111 Free PMC article. Review.
-
Upstream regulators and downstream effectors of NADPH oxidases as novel therapeutic targets for diabetic kidney disease.Mol Cells. 2015 Apr;38(4):285-96. doi: 10.14348/molcells.2015.0010. Epub 2015 Mar 31. Mol Cells. 2015. PMID: 25824546 Free PMC article. Review.
References
-
- Towler MC, Hardie DG. AMP-activated protein kinase in metabolic control and insulin signaling. Circ Res. 2007;100:328–341. - PubMed
-
- Viollet B, Andreelli F, Jorgensen SB, Perrin C, Geloen A, Flamez D, Mu J, Lenzner C, Baud O, Bennoun M, Gomas E, Nicolas G, Wojtaszewski JF, Kahn A, Carling D, Schuit FC, Birnbaum MJ, Richter EA, Burcelin R, Vaulont S. The AMP-activated protein kinase alpha2 catalytic subunit controls whole-body insulin sensitivity. J Clin Invest. 2003;111:91–98. - PMC - PubMed
-
- Zarrinpashneh E, Carjaval K, Beauloye C, Ginion A, Mateo P, Pouleur AC, Horman S, Vaulont S, Hoerter J, Viollet B, Hue L, Vanoverschelde JL, Bertrand L. Role of the alpha2-isoform of AMP-activated protein kinase in the metabolic response of the heart to no-flow ischemia. Am J Physiol Heart Circ Physiol. 2006;291:H2875–2883. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
