

Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I
- PMID: 21194679
- PMCID: PMC3014370
- DOI: 10.1016/j.ajhg.2010.12.003
Targeted high-throughput sequencing identifies mutations in atlastin-1 as a cause of hereditary sensory neuropathy type I
Abstract
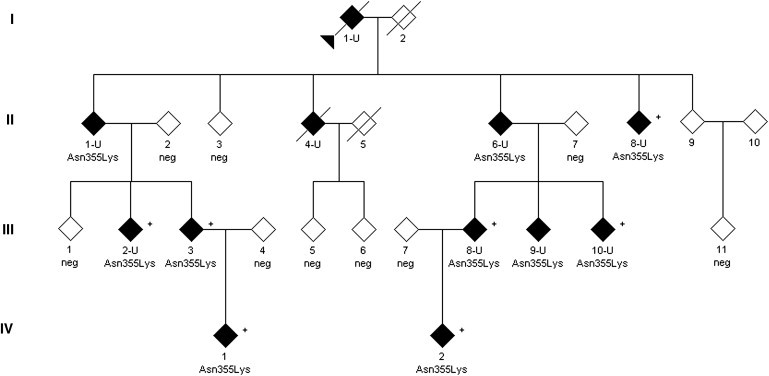
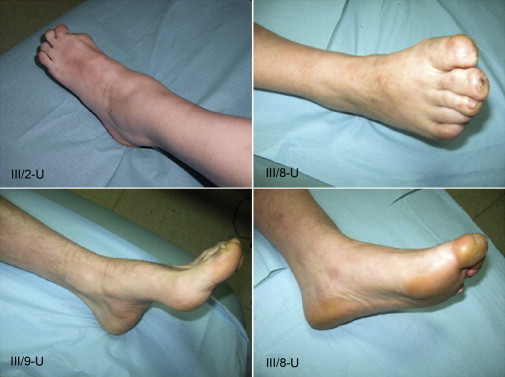
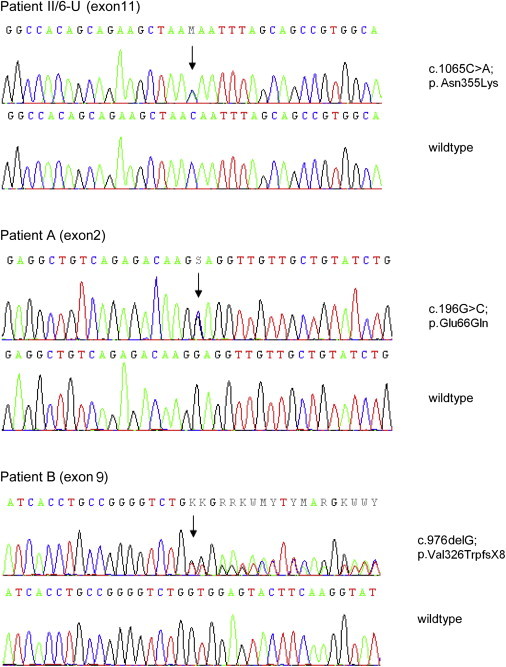
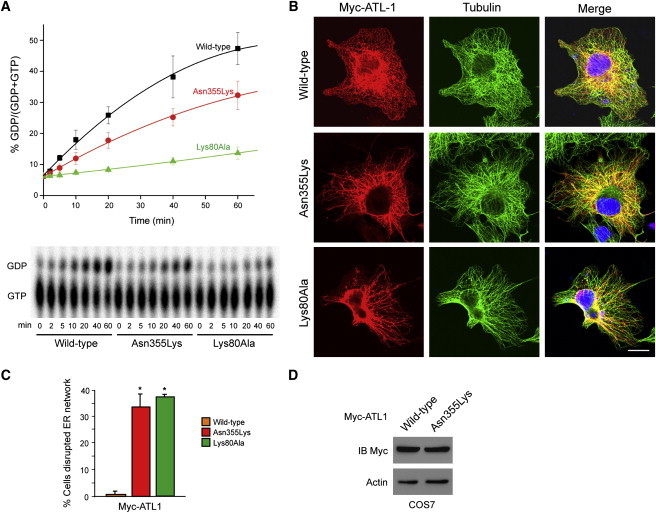
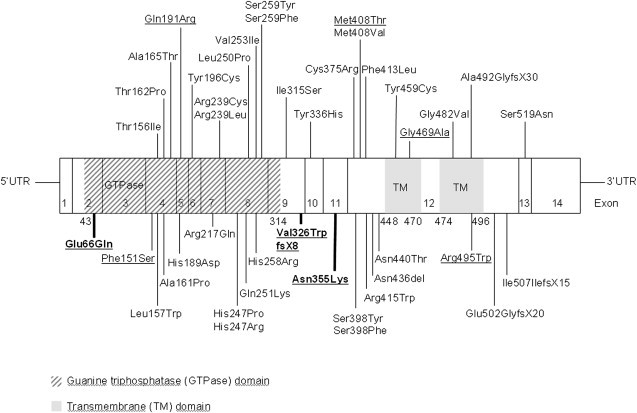
Hereditary sensory neuropathy type I (HSN I) is an axonal form of autosomal-dominant hereditary motor and sensory neuropathy distinguished by prominent sensory loss that leads to painless injuries. Unrecognized, these can result in delayed wound healing and osteomyelitis, necessitating distal amputations. To elucidate the genetic basis of an HSN I subtype in a family in which mutations in the few known HSN I genes had been excluded, we employed massive parallel exon sequencing of the 14.3 Mb disease interval on chromosome 14q. We detected a missense mutation (c.1065C>A, p.Asn355Lys) in atlastin-1 (ATL1), a gene that is known to be mutated in early-onset hereditary spastic paraplegia SPG3A and that encodes the large dynamin-related GTPase atlastin-1. The mutant protein exhibited reduced GTPase activity and prominently disrupted ER network morphology when expressed in COS7 cells, strongly supporting pathogenicity. An expanded screen in 115 additional HSN I patients identified two further dominant ATL1 mutations (c.196G>C [p.Glu66Gln] and c.976 delG [p.Val326TrpfsX8]). This study highlights an unexpected major role for atlastin-1 in the function of sensory neurons and identifies HSN I and SPG3A as allelic disorders.
Figures
Similar articles
-
Sensory neuropathy with bone destruction due to a mutation in the membrane-shaping atlastin GTPase 3.Brain. 2014 Mar;137(Pt 3):683-92. doi: 10.1093/brain/awt357. Epub 2014 Jan 22. Brain. 2014. PMID: 24459106
-
The N355K atlastin 1 mutation is associated with hereditary sensory neuropathy and pyramidal tract features.Eur J Neurol. 2012 Jul;19(7):992-8. doi: 10.1111/j.1468-1331.2012.03665.x. Epub 2012 Feb 16. Eur J Neurol. 2012. PMID: 22340599
-
Novel mutation in the SPG3A gene in an African American family with an early onset of hereditary spastic paraplegia.Arch Neurol. 2004 Oct;61(10):1600-3. doi: 10.1001/archneur.61.10.1600. Arch Neurol. 2004. PMID: 15477516
-
Autosomal dominant inherited neuropathies with prominent sensory loss and mutilations: a review.Arch Neurol. 2003 Mar;60(3):329-34. doi: 10.1001/archneur.60.3.329. Arch Neurol. 2003. PMID: 12633143 Review.
-
Hereditary sensory neuropathies.Drugs Today (Barc). 2004 May;40(5):385-94. doi: 10.1358/dot.2004.40.5.850487. Drugs Today (Barc). 2004. PMID: 15319794 Review.
Cited by
-
Inherited neuropathies: clinical overview and update.Muscle Nerve. 2013 Oct;48(4):604-22. doi: 10.1002/mus.23775. Epub 2013 Jun 26. Muscle Nerve. 2013. PMID: 23801417 Free PMC article. Review.
-
Cellular pathways of hereditary spastic paraplegia.Annu Rev Neurosci. 2012;35:25-47. doi: 10.1146/annurev-neuro-062111-150400. Epub 2012 Apr 20. Annu Rev Neurosci. 2012. PMID: 22540978 Free PMC article. Review.
-
Genetic landscape of congenital insensitivity to pain and hereditary sensory and autonomic neuropathies.Brain. 2023 Dec 1;146(12):4880-4890. doi: 10.1093/brain/awad328. Brain. 2023. PMID: 37769650 Free PMC article.
-
An Update on the Hereditary Spastic Paraplegias: New Genes and New Disease Models.Mov Disord Clin Pract. 2015 Jun 2;2(3):213-223. doi: 10.1002/mdc3.12184. eCollection 2015 Sep. Mov Disord Clin Pract. 2015. PMID: 30838228 Free PMC article. Review.
-
Autophagy in major human diseases.EMBO J. 2021 Oct 1;40(19):e108863. doi: 10.15252/embj.2021108863. Epub 2021 Aug 30. EMBO J. 2021. PMID: 34459017 Free PMC article. Review.
References
-
- Rehman A.U., Morell R.J., Belyantseva I.A., Khan S.Y., Boger E.T., Shahzad M., Ahmed Z.M., Riazuddin S., Khan S.N., Riazuddin S. Targeted capture and next-generation sequencing identifies C9orf75, encoding taperin, as the mutated gene in nonsyndromic deafness DFNB79. Am. J. Hum. Genet. 2010;86:378–388. - PMC - PubMed
-
- Nikopoulos K., Gilissen C., Hoischen A., van Nouhuys C.E., Boonstra F.N., Blokland E.A.W., Arts P., Wieskamp N., Strom T.M., Ayuso C. Next-generation sequencing of a 40 Mb linkage interval reveals TSPAN12 mutations in patients with familial exudative vitreoretinopathy. Am. J. Hum. Genet. 2010;86:240–247. - PMC - PubMed
-
- Nicholson G.A. Hereditary sensory neuropathy I. In: Pagon R.A., Bird T.C., Stephens K., editors. GeneReviews. University of Washington; Seattle, WA: 2010. PMID: 20301564. - PubMed
-
- Dawkins J.L., Hulme D.J., Brahmbhatt S.B., Auer-Grumbach M., Nicholson G.A. Mutations in SPTLC1, encoding serine palmitoyltransferase, long chain base subunit-1, cause hereditary sensory neuropathy type I. Nat. Genet. 2001;27:309–312. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
