

PKR-dependent CHOP induction limits hyperoxia-induced lung injury
- PMID: 21186267
- PMCID: PMC3064291
- DOI: 10.1152/ajplung.00166.2010
PKR-dependent CHOP induction limits hyperoxia-induced lung injury
Abstract
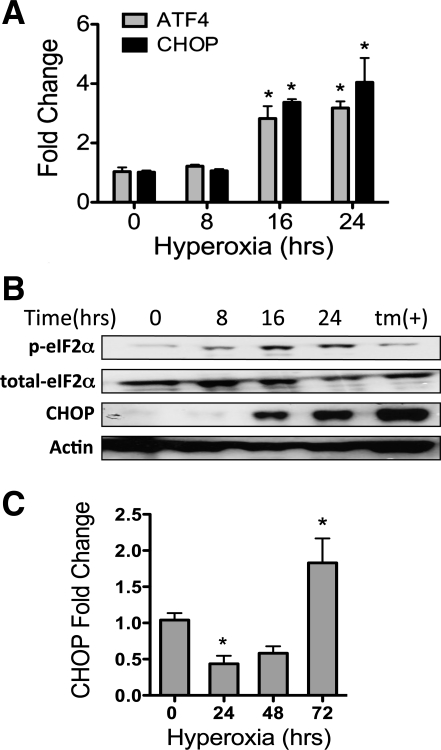
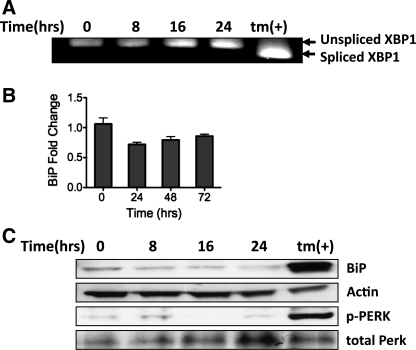
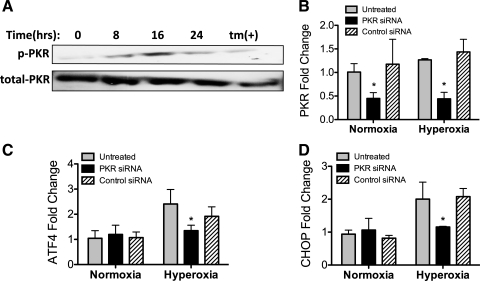
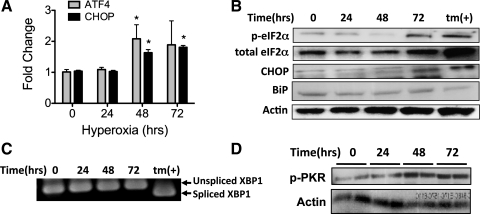
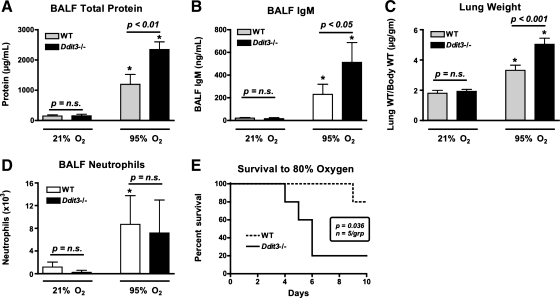
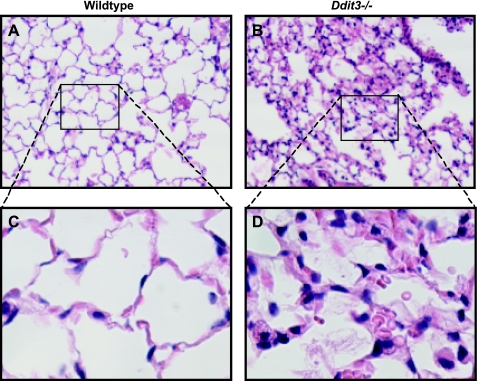
Supplemental O(2) is commonly employed in patients with respiratory failure; however, hyperoxia is also a potential contributor to lung injury. In animal models, hyperoxia causes oxidative stress in the lungs, resulting in increased inflammation, edema, and permeability. We hypothesized that oxidative stress from prolonged hyperoxia leads to endoplasmic reticulum (ER) stress, resulting in activation of the unfolded protein response (UPR) and induction of CCAAT enhancer-binding protein homologous protein (CHOP), a transcription factor associated with cell death in the setting of persistent ER stress. To test this hypothesis, we exposed the mouse lung epithelial cell line MLE-12 to 95% O(2) for 8-24 h and evaluated for evidence of UPR induction and CHOP induction. Hyperoxia caused increased CHOP expression without other evidence of UPR activation. Because CHOP expression is preceded by phosphorylation of the α-subunit of the eukaryotic initiation factor-2 (eIF2α), we evaluated the role of double-stranded RNA-activated protein kinase (PKR), a non-UPR-associated eIF2α kinase. Hyperoxia caused PKR phosphorylation, and RNA interference knockdown of PKR attenuated hyperoxia-induced CHOP expression. In vivo, hyperoxia induced PKR phosphorylation and CHOP expression in the lungs without other biochemical evidence for ER stress. Additionally, Ddit3(-/-) (CHOP-null) mice had increased lung edema and permeability, indicating a previously unknown protective role for CHOP after prolonged hyperoxia. We conclude that hyperoxia increases CHOP expression via an ER stress-independent, PKR-dependent pathway and that increased CHOP expression protects against hyperoxia-induced lung injury.
Figures
Similar articles
-
The double-strand RNA-dependent protein kinase PKR plays a significant role in a sustained ER stress-induced apoptosis.FEBS Lett. 2007 Sep 4;581(22):4325-32. doi: 10.1016/j.febslet.2007.08.001. Epub 2007 Aug 14. FEBS Lett. 2007. PMID: 17716668
-
Cytokine-induced beta-cell death is independent of endoplasmic reticulum stress signaling.Diabetes. 2008 Nov;57(11):3034-44. doi: 10.2337/db07-1802. Epub 2008 Jun 30. Diabetes. 2008. PMID: 18591394 Free PMC article.
-
Localized hypoxia links ER stress to lung fibrosis through induction of C/EBP homologous protein.JCI Insight. 2018 Aug 23;3(16):e99543. doi: 10.1172/jci.insight.99543. eCollection 2018 Aug 23. JCI Insight. 2018. PMID: 30135303 Free PMC article.
-
The Role of the PERK/eIF2α/ATF4/CHOP Signaling Pathway in Tumor Progression During Endoplasmic Reticulum Stress.Curr Mol Med. 2016;16(6):533-44. doi: 10.2174/1566524016666160523143937. Curr Mol Med. 2016. PMID: 27211800 Free PMC article. Review.
-
The unfolded protein response is required to maintain the integrity of the endoplasmic reticulum, prevent oxidative stress and preserve differentiation in β-cells.Diabetes Obes Metab. 2010 Oct;12 Suppl 2(Suppl. 2):99-107. doi: 10.1111/j.1463-1326.2010.01281.x. Diabetes Obes Metab. 2010. PMID: 21029306 Free PMC article. Review.
Cited by
-
Superoxide dismutase 3 dysregulation in a murine model of neonatal lung injury.Am J Respir Cell Mol Biol. 2014 Sep;51(3):380-90. doi: 10.1165/rcmb.2013-0043OC. Am J Respir Cell Mol Biol. 2014. PMID: 24673633 Free PMC article.
-
Expression of Stress-Induced Genes in Bronchoalveolar Lavage Cells and Lung Fibroblasts from Healthy and COPD Subjects.Int J Mol Sci. 2024 Jun 15;25(12):6600. doi: 10.3390/ijms25126600. Int J Mol Sci. 2024. PMID: 38928305 Free PMC article.
-
Ischemia/reperfusion-induced CHOP expression promotes apoptosis and impairs renal function recovery: the role of acidosis and GPR4.PLoS One. 2014 Oct 24;9(10):e110944. doi: 10.1371/journal.pone.0110944. eCollection 2014. PLoS One. 2014. PMID: 25343248 Free PMC article.
-
Transcription Factor C/EBP Homologous Protein in Health and Diseases.Front Immunol. 2017 Nov 27;8:1612. doi: 10.3389/fimmu.2017.01612. eCollection 2017. Front Immunol. 2017. PMID: 29230213 Free PMC article. Review.
-
Androgen signaling promotes translation of TMEFF2 in prostate cancer cells via phosphorylation of the α subunit of the translation initiation factor 2.PLoS One. 2013;8(2):e55257. doi: 10.1371/journal.pone.0055257. Epub 2013 Feb 6. PLoS One. 2013. PMID: 23405127 Free PMC article.
References
-
- Altemeier WA, Sinclair SE. Hyperoxia in the intensive care unit: why more is not always better. Curr Opin Crit Care 13: 73–78, 2007 - PubMed
-
- Beck KC, Vettermann J, Rehder K. Gas exchange in dogs in the prone and supine positions. J Appl Physiol 72: 2292–2297, 1992 - PubMed
-
- Bernales S, Papa FR, Walter P. Intracellular signaling by the unfolded protein response. Annu Rev Cell Dev Biol 22: 487–508, 2006 - PubMed
-
- Bhandari V, Elias JA. The role of angiopoietin 2 in hyperoxia-induced acute lung injury. Cell Cycle 6: 1049–1052, 2007 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials