

Alternate states of proteins revealed by detailed energy landscape mapping
- PMID: 21073878
- PMCID: PMC3046547
- DOI: 10.1016/j.jmb.2010.11.008
Alternate states of proteins revealed by detailed energy landscape mapping
Abstract
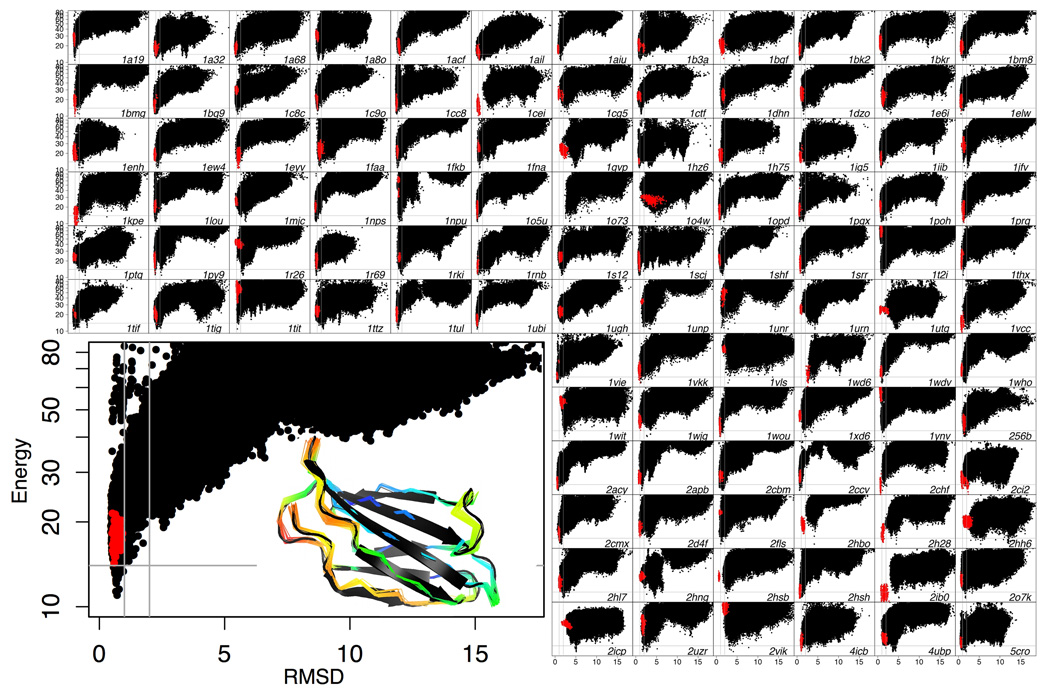
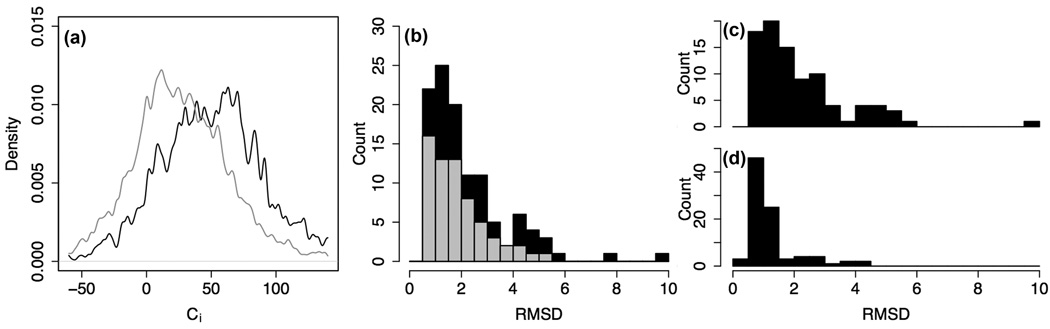
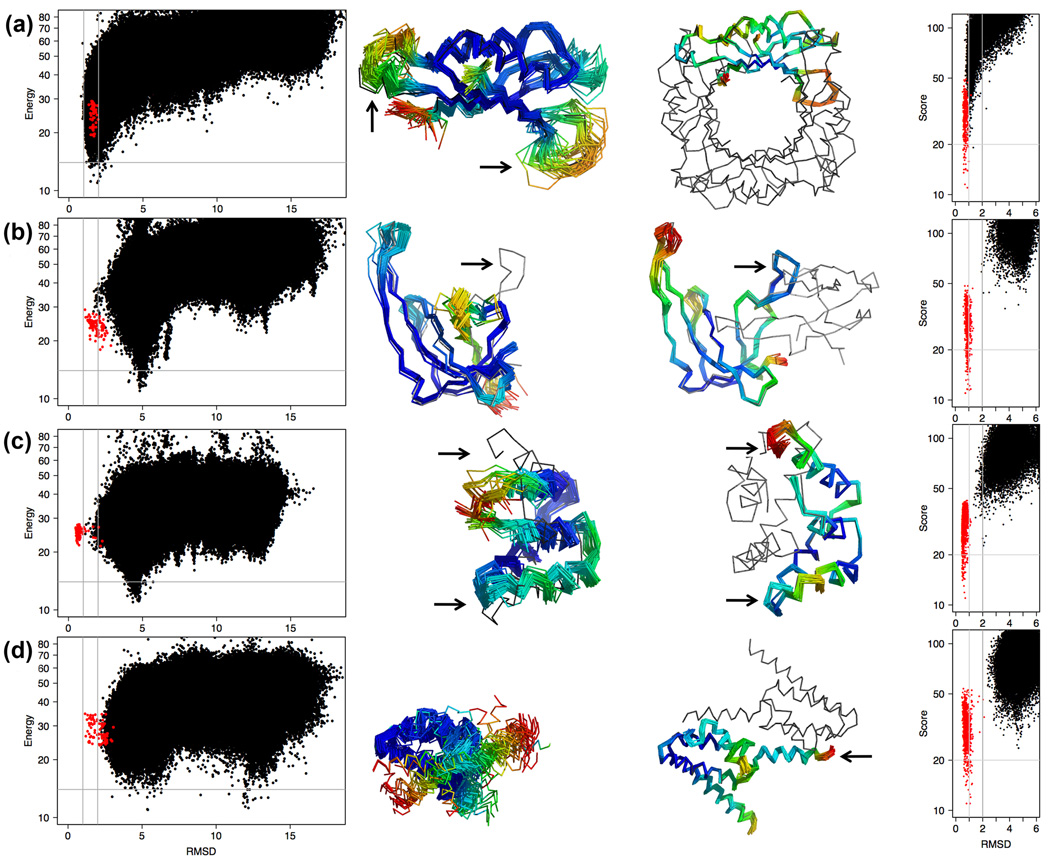
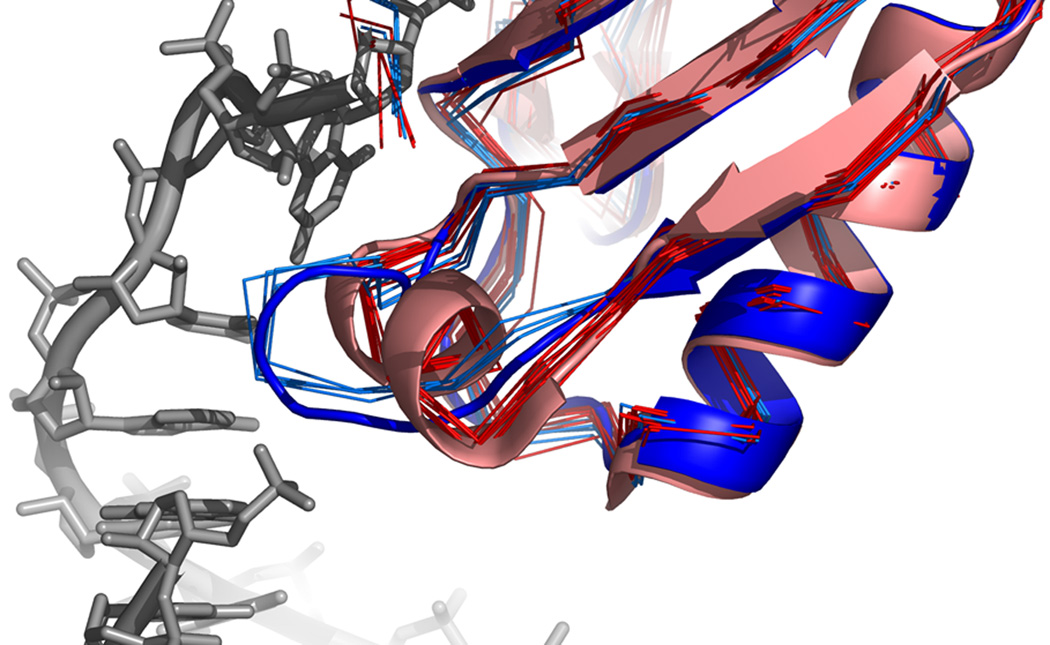
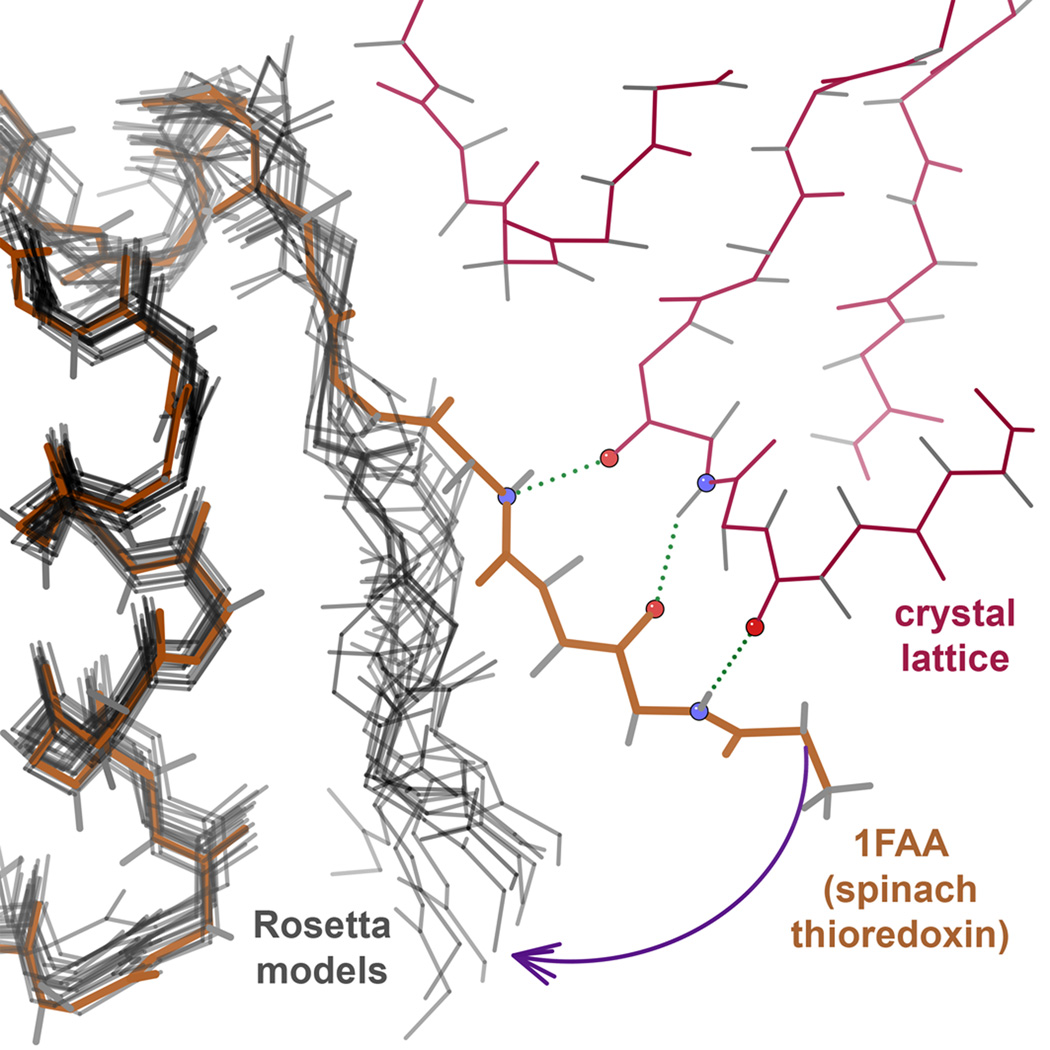
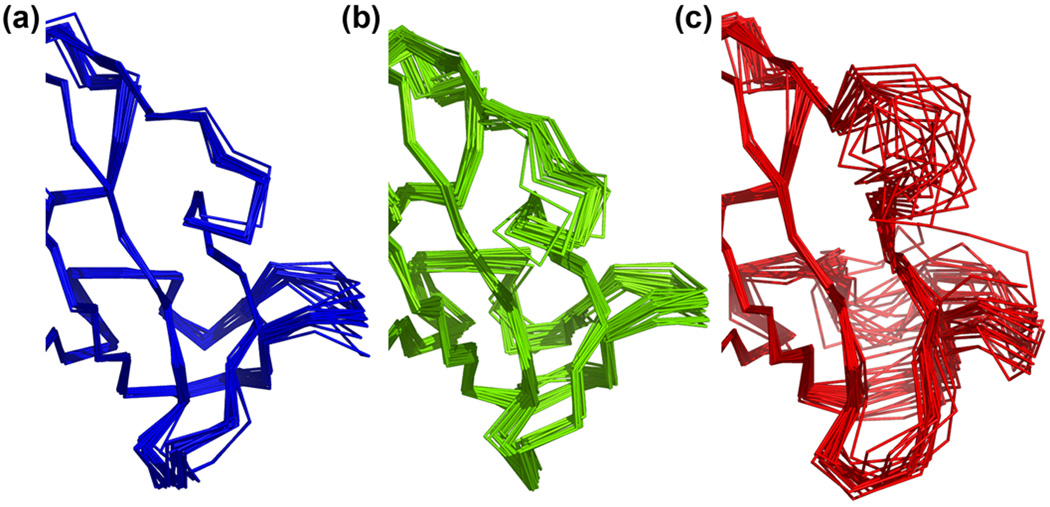
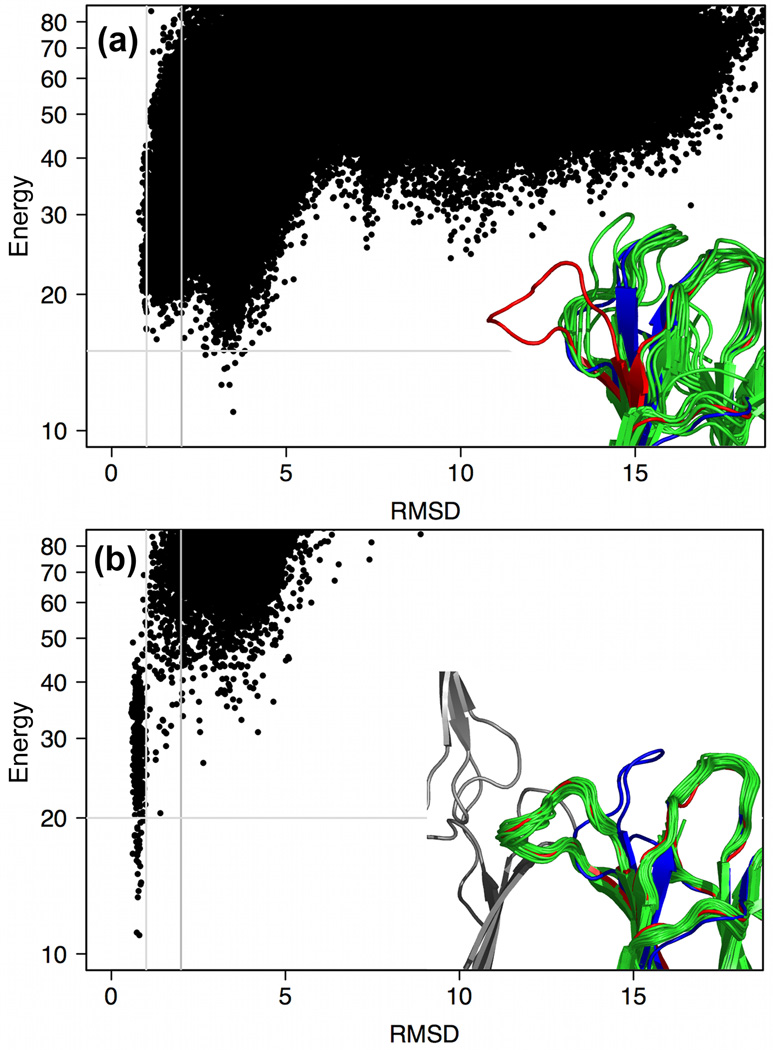
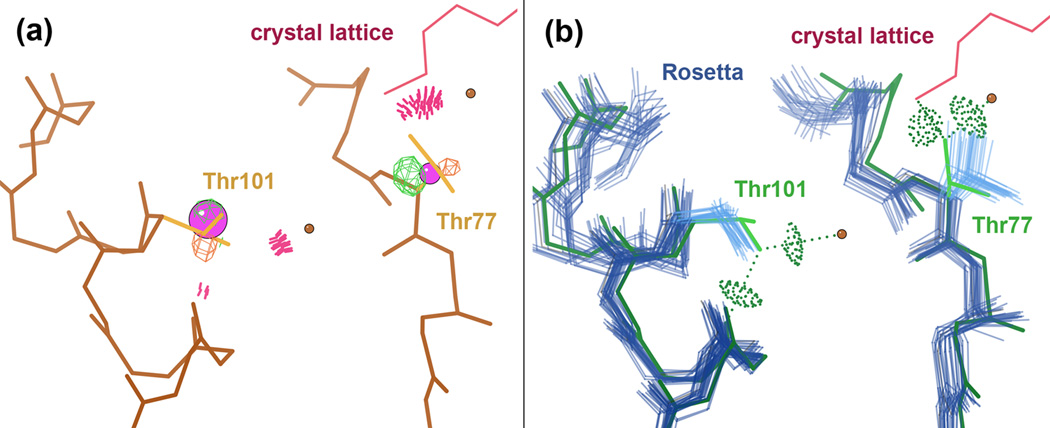
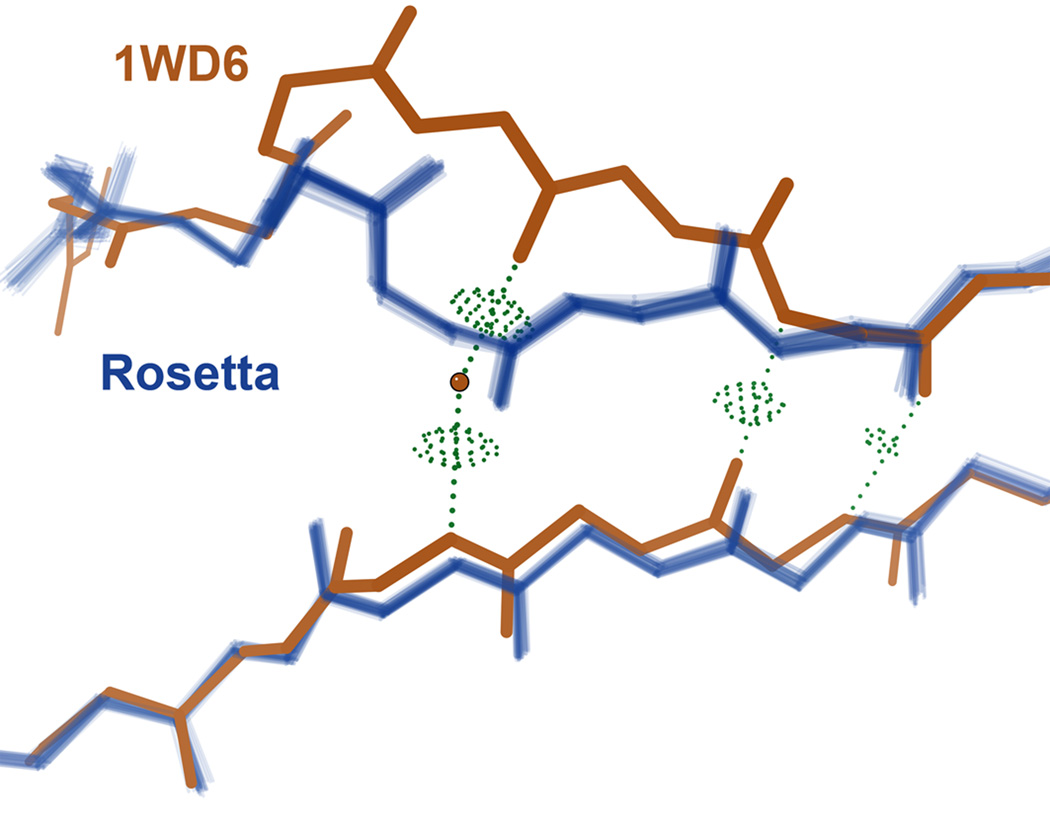
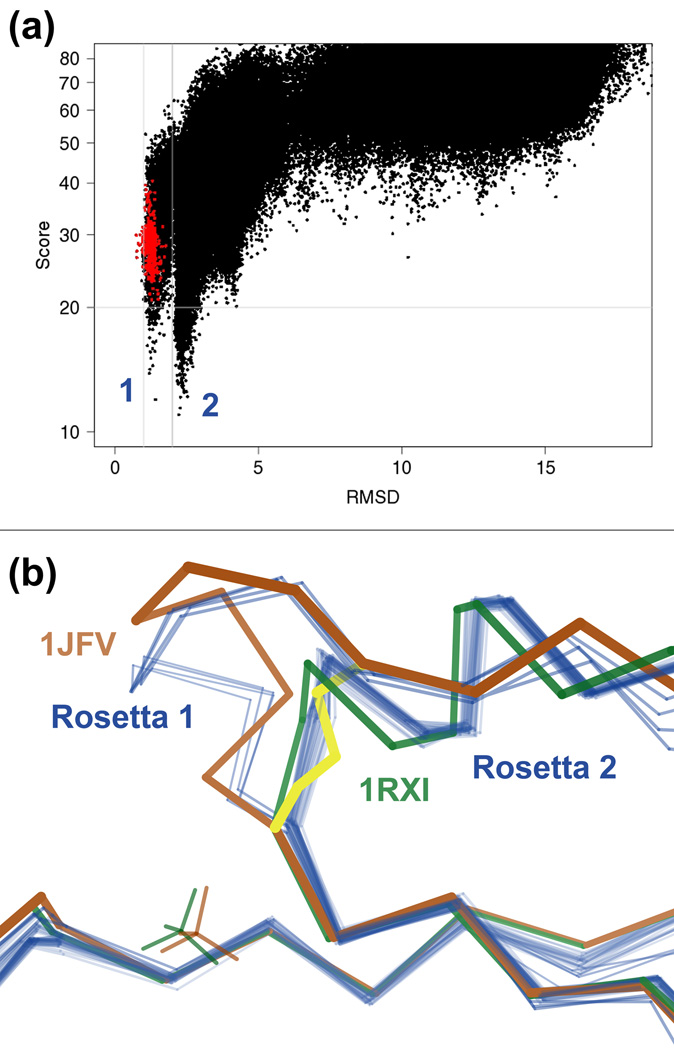
What conformations do protein molecules populate in solution? Crystallography provides a high-resolution description of protein structure in the crystal environment, while NMR describes structure in solution but using less data. NMR structures display more variability, but is this because crystal contacts are absent or because of fewer data constraints? Here we report unexpected insight into this issue obtained through analysis of detailed protein energy landscapes generated by large-scale, native-enhanced sampling of conformational space with Rosetta@home for 111 protein domains. In the absence of tightly associating binding partners or ligands, the lowest-energy Rosetta models were nearly all <2.5 Å C(α)RMSD from the experimental structure; this result demonstrates that structure prediction accuracy for globular proteins is limited mainly by the ability to sample close to the native structure. While the lowest-energy models are similar to deposited structures, they are not identical; the largest deviations are most often in regions involved in ligand, quaternary, or crystal contacts. For ligand binding proteins, the low energy models may resemble the apo structures, and for oligomeric proteins, the monomeric assembly intermediates. The deviations between the low energy models and crystal structures largely disappear when landscapes are computed in the context of the crystal lattice or multimer. The computed low-energy ensembles, with tight crystal-structure-like packing in the core, but more NMR-structure-like variability in loops, may in some cases resemble the native state ensembles of proteins better than individual crystal or NMR structures, and can suggest experimentally testable hypotheses relating alternative states and structural heterogeneity to function.
Copyright © 2010 Elsevier Ltd. All rights reserved.
Figures
Similar articles
-
Improving NMR protein structure quality by Rosetta refinement: a molecular replacement study.Proteins. 2009 Apr;75(1):147-67. doi: 10.1002/prot.22229. Proteins. 2009. PMID: 18816799 Free PMC article.
-
Variability of conformations at crystal contacts in BPTI represent true low-energy structures: correspondence among lattice packing and molecular dynamics structures.Proteins. 1992 Sep;14(1):65-74. doi: 10.1002/prot.340140108. Proteins. 1992. PMID: 1384033
-
Protein NMR structures refined with Rosetta have higher accuracy relative to corresponding X-ray crystal structures.J Am Chem Soc. 2014 Feb 5;136(5):1893-906. doi: 10.1021/ja409845w. Epub 2014 Jan 23. J Am Chem Soc. 2014. PMID: 24392845 Free PMC article.
-
The folding energy landscape and free energy excitations of cytochrome c.Acc Chem Res. 2010 May 18;43(5):652-60. doi: 10.1021/ar9002703. Acc Chem Res. 2010. PMID: 20143816 Free PMC article. Review.
-
From crystal structure prediction to polymorph prediction: interpreting the crystal energy landscape.Phys Chem Chem Phys. 2008 Apr 21;10(15):1996-2009. doi: 10.1039/b719351c. Epub 2008 Feb 19. Phys Chem Chem Phys. 2008. PMID: 18688351 Review.
Cited by
-
Tarantula toxins use common surfaces for interacting with Kv and ASIC ion channels.Elife. 2015 May 7;4:e06774. doi: 10.7554/eLife.06774. Elife. 2015. PMID: 25948544 Free PMC article.
-
Photoswitchable affinity reagents: Computational design and efficient red-light switching.ChemPhotoChem. 2019 Jun;3(6):431-440. doi: 10.1002/cptc.201900016. Epub 2019 Mar 4. ChemPhotoChem. 2019. PMID: 32856001 Free PMC article.
-
Structure of the basal components of a bacterial transporter.Proc Natl Acad Sci U S A. 2012 Apr 3;109(14):5446-51. doi: 10.1073/pnas.1120113109. Epub 2012 Mar 19. Proc Natl Acad Sci U S A. 2012. PMID: 22431613 Free PMC article.
-
Quantitative structural assessment of graded receptor agonism.Proc Natl Acad Sci U S A. 2019 Oct 29;116(44):22179-22188. doi: 10.1073/pnas.1909016116. Epub 2019 Oct 14. Proc Natl Acad Sci U S A. 2019. PMID: 31611383 Free PMC article.
-
Reconfigurable asymmetric protein assemblies through implicit negative design.Science. 2022 Jan 21;375(6578):eabj7662. doi: 10.1126/science.abj7662. Epub 2022 Jan 21. Science. 2022. PMID: 35050655 Free PMC article.
References
-
- Bradley P, Misura KMS, Baker D. Toward high-resolution de novo structure prediction for small proteins. Science. 2005;309:1868–1871. - PubMed
-
- Das R, et al. Structure prediction for CASP7 targets using extensive all-atom refinement with Rosetta@HOME. Proteins: Struc Func Bioinf. 2007;69 Suppl 8:118–128. - PubMed
-
- Kuhlman B, et al. Design of a Novel Globular Protein Fold with Atomic-Level Accuracy. Science. 2003;302:1364–1368. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
