

The honey bee epigenomes: differential methylation of brain DNA in queens and workers
- PMID: 21072239
- PMCID: PMC2970541
- DOI: 10.1371/journal.pbio.1000506
The honey bee epigenomes: differential methylation of brain DNA in queens and workers
Erratum in
- PLoS Biol. 2011;9(1). doi: 10.1371/annotation/2db9ee19-faa4-43f2-af7a-c8aeacca8037
Abstract
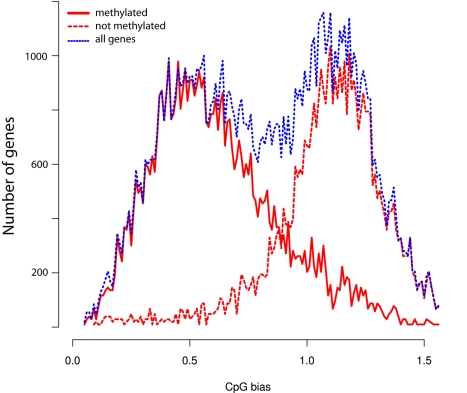
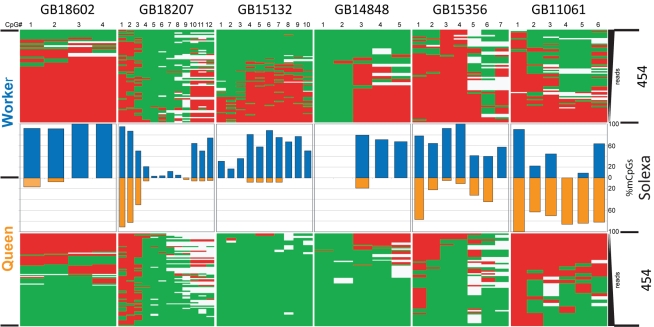
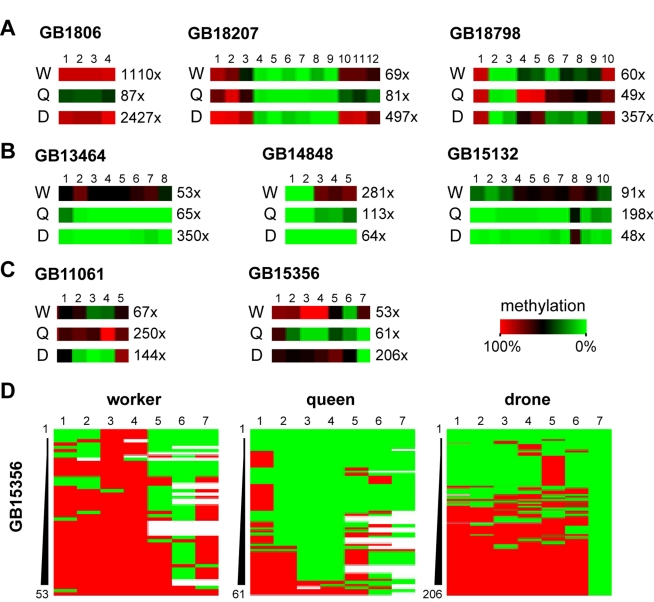
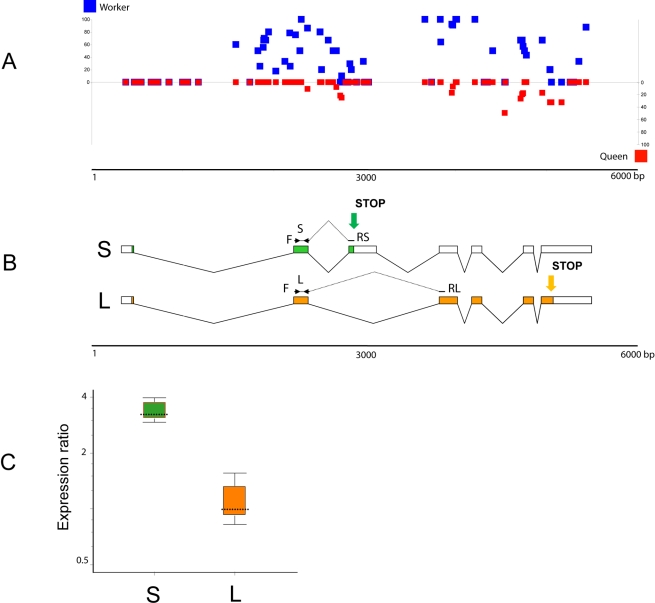
In honey bees (Apis mellifera) the behaviorally and reproductively distinct queen and worker female castes derive from the same genome as a result of differential intake of royal jelly and are implemented in concert with DNA methylation. To determine if these very different diet-controlled phenotypes correlate with unique brain methylomes, we conducted a study to determine the methyl cytosine (mC) distribution in the brains of queens and workers at single-base-pair resolution using shotgun bisulfite sequencing technology. The whole-genome sequencing was validated by deep 454 sequencing of selected amplicons representing eight methylated genes. We found that nearly all mCs are located in CpG dinucleotides in the exons of 5,854 genes showing greater sequence conservation than non-methylated genes. Over 550 genes show significant methylation differences between queens and workers, revealing the intricate dynamics of methylation patterns. The distinctiveness of the differentially methylated genes is underscored by their intermediate CpG densities relative to drastically CpG-depleted methylated genes and to CpG-richer non-methylated genes. We find a strong correlation between methylation patterns and splicing sites including those that have the potential to generate alternative exons. We validate our genome-wide analyses by a detailed examination of two transcript variants encoded by one of the differentially methylated genes. The link between methylation and splicing is further supported by the differential methylation of genes belonging to the histone gene family. We propose that modulation of alternative splicing is one mechanism by which DNA methylation could be linked to gene regulation in the honey bee. Our study describes a level of molecular diversity previously unknown in honey bees that might be important for generating phenotypic flexibility not only during development but also in the adult post-mitotic brain.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
DNA methylation dynamics, metabolic fluxes, gene splicing, and alternative phenotypes in honey bees.Proc Natl Acad Sci U S A. 2012 Mar 27;109(13):4968-73. doi: 10.1073/pnas.1202392109. Epub 2012 Mar 13. Proc Natl Acad Sci U S A. 2012. PMID: 22416128 Free PMC article.
-
Roles of DNA Methylation in Color Alternation of Eastern Honey Bees (Apis cerana) Induced by the Royal Jelly of Western Honey Bees (Apis mellifera).Int J Mol Sci. 2024 Mar 16;25(6):3368. doi: 10.3390/ijms25063368. Int J Mol Sci. 2024. PMID: 38542342 Free PMC article.
-
Intronic non-CG DNA hydroxymethylation and alternative mRNA splicing in honey bees.BMC Genomics. 2013 Sep 30;14:666. doi: 10.1186/1471-2164-14-666. BMC Genomics. 2013. PMID: 24079845 Free PMC article.
-
DNA Methylation and Gene Regulation in Honeybees: From Genome-Wide Analyses to Obligatory Epialleles.Adv Exp Med Biol. 2016;945:193-211. doi: 10.1007/978-3-319-43624-1_9. Adv Exp Med Biol. 2016. PMID: 27826840 Review.
-
Cytosine modifications in the honey bee (Apis mellifera) worker genome.Front Genet. 2015 Feb 6;6:8. doi: 10.3389/fgene.2015.00008. eCollection 2015. Front Genet. 2015. PMID: 25705215 Free PMC article. Review.
Cited by
-
DNA methylation and chromatin organization in insects: insights from the Ant Camponotus floridanus.Genome Biol Evol. 2015 Feb 26;7(4):931-42. doi: 10.1093/gbe/evv039. Genome Biol Evol. 2015. PMID: 25724207 Free PMC article.
-
Social parasitism and the molecular basis of phenotypic evolution.Front Genet. 2015 Feb 18;6:32. doi: 10.3389/fgene.2015.00032. eCollection 2015. Front Genet. 2015. PMID: 25741361 Free PMC article.
-
DNA methylation mediates the discriminatory power of associative long-term memory in honeybees.PLoS One. 2012;7(6):e39349. doi: 10.1371/journal.pone.0039349. Epub 2012 Jun 18. PLoS One. 2012. PMID: 22724000 Free PMC article.
-
Evolution of recombination and genome structure in eusocial insects.Commun Integr Biol. 2013 Mar 1;6(2):e22919. doi: 10.4161/cib.22919. Commun Integr Biol. 2013. PMID: 23748924 Free PMC article.
-
A possible role of DNA methylation in functional divergence of a fast evolving duplicate gene encoding odorant binding protein 11 in the honeybee.Proc Biol Sci. 2016 Jun 29;283(1833):20160558. doi: 10.1098/rspb.2016.0558. Proc Biol Sci. 2016. PMID: 27358363 Free PMC article.
References
-
- Maleszka R. Epigenetic integration of environmental and genomic signals in honey bees: the critical interplay of nutritional, brain and reproductive networks. Epigenetics. 2008;3:188–192. - PubMed
-
- Kucharski R, Maleszka J, Foret S, Maleszka R. Nutritional control of reproductive status in honey bees via DNA methylation. Science. 2008;319:1827–1830. - PubMed
-
- Zemach A, McDaniel I. E, Silva P, Zilberman D. Genome-wide evolutionary analysis of eukaryotic DNA methylation. Science. 2010;328:916–919. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
