

Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression
- PMID: 21071446
- PMCID: PMC3023525
- DOI: 10.1074/jbc.M110.169839
Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression
Erratum in
-
Fractalkine attenuates excito-neurotoxicity via microglial clearance of damaged neurons and antioxidant enzyme heme oxygenase-1 expression.J Biol Chem. 2016 Jul 1;291(27):14388. doi: 10.1074/jbc.A110.169839. J Biol Chem. 2016. PMID: 27371564 Free PMC article. No abstract available.
Abstract
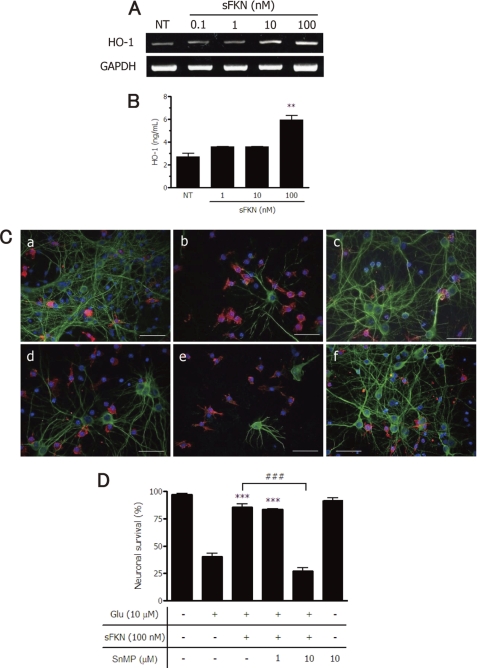
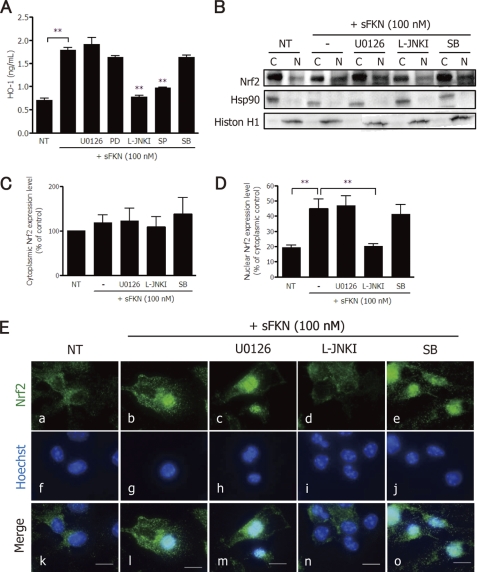
Glutamate-induced excito-neurotoxicity likely contributes to non-cell autonomous neuronal death in neurodegenerative diseases. Microglial clearance of dying neurons and associated debris is essential to maintain healthy neural networks in the central nervous system. In fact, the functions of microglia are regulated by various signaling molecules that are produced as neurons degenerate. Here, we show that the soluble CX3C chemokine fractalkine (sFKN), which is secreted from neurons that have been damaged by glutamate, promotes microglial phagocytosis of neuronal debris through release of milk fat globule-EGF factor 8, a mediator of apoptotic cell clearance. In addition, sFKN induces the expression of the antioxidant enzyme heme oxygenase-1 (HO-1) in microglia in the absence of neurotoxic molecule production, including NO, TNF, and glutamate. sFKN treatment of primary neuron-microglia co-cultures significantly attenuated glutamate-induced neuronal cell death. Using several specific MAPK inhibitors, we found that sFKN-induced heme oxygenase-1 expression was primarily mediated by activation of JNK and nuclear factor erythroid 2-related factor 2. These results suggest that sFKN secreted from glutamate-damaged neurons provides both phagocytotic and neuroprotective signals.
Figures






Similar articles
-
The neuroprotective effects of milk fat globule-EGF factor 8 against oligomeric amyloid β toxicity.J Neuroinflammation. 2012 Jun 28;9:148. doi: 10.1186/1742-2094-9-148. J Neuroinflammation. 2012. PMID: 22742657 Free PMC article.
-
MFG-E8 regulates microglial phagocytosis of apoptotic neurons.J Neuroimmune Pharmacol. 2008 Dec;3(4):246-56. doi: 10.1007/s11481-008-9118-2. Epub 2008 Aug 1. J Neuroimmune Pharmacol. 2008. PMID: 18670887 Free PMC article.
-
Production and neuroprotective functions of fractalkine in the central nervous system.Brain Res. 2003 Jul 25;979(1-2):65-70. doi: 10.1016/s0006-8993(03)02867-1. Brain Res. 2003. PMID: 12850572
-
[Neuronal dysfunction in multiple sclerosis].Rinsho Shinkeigaku. 2014;54(12):1066-8. doi: 10.5692/clinicalneurol.54.1066. Rinsho Shinkeigaku. 2014. PMID: 25519963 Review. Japanese.
-
Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes.Int J Mol Sci. 2018 Jan 22;19(1):318. doi: 10.3390/ijms19010318. Int J Mol Sci. 2018. PMID: 29361745 Free PMC article. Review.
Cited by
-
The Emerging Role of the Interplay Among Astrocytes, Microglia, and Neurons in the Hippocampus in Health and Disease.Front Aging Neurosci. 2021 Apr 6;13:651973. doi: 10.3389/fnagi.2021.651973. eCollection 2021. Front Aging Neurosci. 2021. PMID: 33889084 Free PMC article. Review.
-
HIV-associated synaptic degeneration.Mol Brain. 2017 Aug 29;10(1):40. doi: 10.1186/s13041-017-0321-z. Mol Brain. 2017. PMID: 28851400 Free PMC article. Review.
-
Complex molecular and functional outcomes of single versus sequential cytokine stimulation of rat microglia.J Neuroinflammation. 2016 Mar 24;13(1):66. doi: 10.1186/s12974-016-0531-9. J Neuroinflammation. 2016. PMID: 27009332 Free PMC article.
-
The neuroprotective effects of milk fat globule-EGF factor 8 against oligomeric amyloid β toxicity.J Neuroinflammation. 2012 Jun 28;9:148. doi: 10.1186/1742-2094-9-148. J Neuroinflammation. 2012. PMID: 22742657 Free PMC article.
-
Neurosteroid [3α,5α]-3-Hydroxy-pregnan-20-one Enhances the CX3CL1-CX3CR1 Pathway in the Brain of Alcohol-Preferring Rats with Sex-Specificity.Life (Basel). 2024 Jul 9;14(7):860. doi: 10.3390/life14070860. Life (Basel). 2024. PMID: 39063614 Free PMC article.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
