

Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs
- PMID: 20956553
- PMCID: PMC3019850
- DOI: 10.1128/MCB.00821-10
Genomic collaboration of estrogen receptor alpha and extracellular signal-regulated kinase 2 in regulating gene and proliferation programs
Abstract
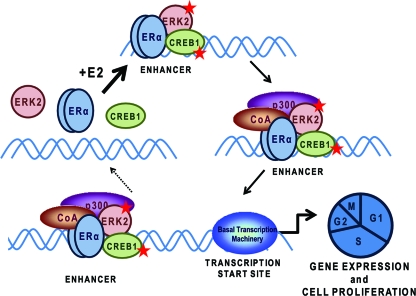
The nuclear hormone receptor, estrogen receptor α (ERα), and mitogen-activated protein kinases (MAPKs) play key roles in hormone-dependent cancers, and yet their interplay and the integration of their signaling inputs remain poorly understood. In these studies, we document that estrogen-occupied ERα activates and interacts with extracellular signal-regulated kinase 2 (ERK2), a downstream effector in the MAPK pathway, resulting in ERK2 and ERα colocalization at chromatin binding sites across the genome of breast cancer cells. This genomic colocalization, predominantly at conserved distal enhancer sites, requires the activation of both ERα and ERK2 and enables ERK2 modulation of estrogen-dependent gene expression and proliferation programs. The ERK2 substrate CREB1 was also activated and recruited to ERK2-bound chromatin following estrogen treatment and found to cooperate with ERα/ERK2 in regulating gene transcription and cell cycle progression. Our study reveals a novel paradigm with convergence of ERK2 and ERα at the chromatin level that positions this kinase to support nuclear receptor activities in crucial and direct ways, a mode of collaboration likely to underlie MAPK regulation of gene expression by other nuclear receptors as well.
Figures





Similar articles
-
A MicroRNA196a2* and TP63 circuit regulated by estrogen receptor-α and ERK2 that controls breast cancer proliferation and invasiveness properties.Horm Cancer. 2013 Apr;4(2):78-91. doi: 10.1007/s12672-012-0129-3. Epub 2012 Dec 19. Horm Cancer. 2013. PMID: 23250869 Free PMC article.
-
Differential utilization of nuclear and extranuclear receptor signaling pathways in the actions of estrogens, SERMs, and a tissue-selective estrogen complex (TSEC).J Steroid Biochem Mol Biol. 2016 Apr;158:198-206. doi: 10.1016/j.jsbmb.2015.12.008. Epub 2015 Dec 12. J Steroid Biochem Mol Biol. 2016. PMID: 26689478
-
Nuclear and extranuclear pathway inputs in the regulation of global gene expression by estrogen receptors.Mol Endocrinol. 2008 Sep;22(9):2116-27. doi: 10.1210/me.2008-0059. Epub 2008 Jul 10. Mol Endocrinol. 2008. PMID: 18617595 Free PMC article.
-
Mechanisms of estrogen receptor-α upregulation in breast cancers.Med Mol Morphol. 2010 Dec;43(4):193-6. doi: 10.1007/s00795-010-0514-3. Epub 2011 Jan 26. Med Mol Morphol. 2010. PMID: 21267694 Review.
-
Hormone-regulated transcriptomes: lessons learned from estrogen signaling pathways in breast cancer cells.Mol Cell Endocrinol. 2014 Jan 25;382(1):652-664. doi: 10.1016/j.mce.2013.06.021. Epub 2013 Jun 28. Mol Cell Endocrinol. 2014. PMID: 23810978 Free PMC article. Review.
Cited by
-
Reprogramming of endothelial gene expression by tamoxifen inhibits angiogenesis and ERα-negative tumor growth.Theranostics. 2024 Jan 1;14(1):249-264. doi: 10.7150/thno.87306. eCollection 2024. Theranostics. 2024. PMID: 38164151 Free PMC article.
-
Sex Differences in Colon Cancer: Genomic and Nongenomic Signalling of Oestrogen.Genes (Basel). 2023 Dec 16;14(12):2225. doi: 10.3390/genes14122225. Genes (Basel). 2023. PMID: 38137047 Free PMC article. Review.
-
A novel selective ERK1/2 inhibitor, Laxiflorin B, targets EGFR mutation subtypes in non-small-cell lung cancer.Acta Pharmacol Sin. 2024 Feb;45(2):422-435. doi: 10.1038/s41401-023-01164-w. Epub 2023 Oct 10. Acta Pharmacol Sin. 2024. PMID: 37816856
-
Gene transcription regulation by ER at the single cell and allele level.Steroids. 2023 Dec;200:109313. doi: 10.1016/j.steroids.2023.109313. Epub 2023 Sep 25. Steroids. 2023. PMID: 37758052
-
Immunolocalization of Cytoplasmic ER in ER-negative Breast Carcinoma as a Potent Favorable Prognostic Predictor.Acta Histochem Cytochem. 2023 Aug 30;56(4):59-66. doi: 10.1267/ahc.23-00016. Epub 2023 Aug 23. Acta Histochem Cytochem. 2023. PMID: 37680573 Free PMC article.
References
-
- Britton, D. J., I. R. Hutcheson, J. M. Knowlden, D. Barrow, M. Giles, R. A. McClelland, J. M. Gee, and R. I. Nicholson. 2006. Bidirectional cross talk between ERα and EGFR signaling pathways regulates tamoxifen-resistant growth. Breast Cancer Res. Treat 96:131-146. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous