

HIV promoter integration site primarily modulates transcriptional burst size rather than frequency
- PMID: 20941390
- PMCID: PMC2947985
- DOI: 10.1371/journal.pcbi.1000952
HIV promoter integration site primarily modulates transcriptional burst size rather than frequency
Abstract
Mammalian gene expression patterns, and their variability across populations of cells, are regulated by factors specific to each gene in concert with its surrounding cellular and genomic environment. Lentiviruses such as HIV integrate their genomes into semi-random genomic locations in the cells they infect, and the resulting viral gene expression provides a natural system to dissect the contributions of genomic environment to transcriptional regulation. Previously, we showed that expression heterogeneity and its modulation by specific host factors at HIV integration sites are key determinants of infected-cell fate and a possible source of latent infections. Here, we assess the integration context dependence of expression heterogeneity from diverse single integrations of a HIV-promoter/GFP-reporter cassette in Jurkat T-cells. Systematically fitting a stochastic model of gene expression to our data reveals an underlying transcriptional dynamic, by which multiple transcripts are produced during short, infrequent bursts, that quantitatively accounts for the wide, highly skewed protein expression distributions observed in each of our clonal cell populations. Interestingly, we find that the size of transcriptional bursts is the primary systematic covariate over integration sites, varying from a few to tens of transcripts across integration sites, and correlating well with mean expression. In contrast, burst frequencies are scattered about a typical value of several per cell-division time and demonstrate little correlation with the clonal means. This pattern of modulation generates consistently noisy distributions over the sampled integration positions, with large expression variability relative to the mean maintained even for the most productive integrations, and could contribute to specifying heterogeneous, integration-site-dependent viral production patterns in HIV-infected cells. Genomic environment thus emerges as a significant control parameter for gene expression variation that may contribute to structuring mammalian genomes, as well as be exploited for survival by integrating viruses.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
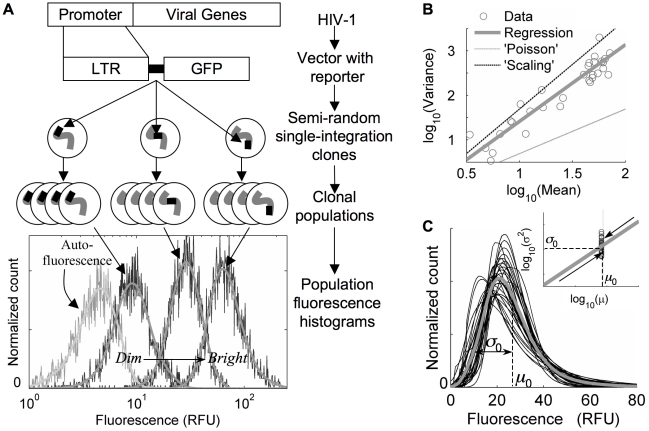
 ;
; 
 (R2 = 0.89, coefficients ±95% confidence). Dashed lines demonstrate Poisson-like scaling (‘Poisson’, α = 1) and over-all distribution scaling (‘Scaling’, α = 2). C) Characteristic distribution shape: Smoothed, autoflorescence-deconvolved histograms were shifted by a constant fluorescence to a fixed mean (specified by the median over the set of integration clones, μ0), and fluorescence values were scaled about that mean according to the variance regression in 1B (inset), by a factor
(R2 = 0.89, coefficients ±95% confidence). Dashed lines demonstrate Poisson-like scaling (‘Poisson’, α = 1) and over-all distribution scaling (‘Scaling’, α = 2). C) Characteristic distribution shape: Smoothed, autoflorescence-deconvolved histograms were shifted by a constant fluorescence to a fixed mean (specified by the median over the set of integration clones, μ0), and fluorescence values were scaled about that mean according to the variance regression in 1B (inset), by a factor  with α = 1.7 (thin curves). The grey curve averages the transformed distributions and represents a ‘typical’ HIV-LTR expression profile that is wide (coefficient of variation = σ0/μ0∼60%) and highly skewed.
with α = 1.7 (thin curves). The grey curve averages the transformed distributions and represents a ‘typical’ HIV-LTR expression profile that is wide (coefficient of variation = σ0/μ0∼60%) and highly skewed.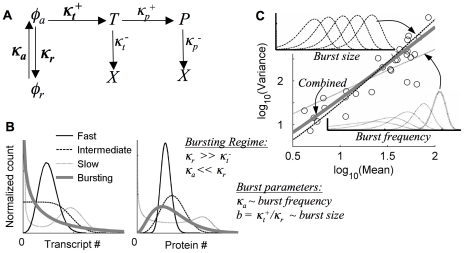
 ), ‘Slow’ (
), ‘Slow’ ( ), ‘Intermediate’ (
), ‘Intermediate’ ( ), ‘Bursting’ (
), ‘Bursting’ ( ). Transcript production rates (κt+) for sample distributions are set to reproduce the same mean number of transcript copies (left) and protein copies (right) at steady state as predicted for the ‘typical’ experimental distribution in Fig. 1C; autoflourescence is not included, and distributions are binned on a linear RFU scale for comparison. C) Distribution shape-variation in the bursting regime: burst-frequency variation (κa) leads to an approximate Poisson-like shape-variation, burst-size (b) yields an approximate distribution-scaling shape variation, and the combination with
). Transcript production rates (κt+) for sample distributions are set to reproduce the same mean number of transcript copies (left) and protein copies (right) at steady state as predicted for the ‘typical’ experimental distribution in Fig. 1C; autoflourescence is not included, and distributions are binned on a linear RFU scale for comparison. C) Distribution shape-variation in the bursting regime: burst-frequency variation (κa) leads to an approximate Poisson-like shape-variation, burst-size (b) yields an approximate distribution-scaling shape variation, and the combination with  (combined) gives a shape-variation most closely resembling the experimental data (compare Fig. 1B). Insets are sample, log-binned distributions with varying burst size or frequency. The fixed parameter in each sample is set to the value that approximately reproduces the ‘typical’ distribution shape in Fig. 1C.
(combined) gives a shape-variation most closely resembling the experimental data (compare Fig. 1B). Insets are sample, log-binned distributions with varying burst size or frequency. The fixed parameter in each sample is set to the value that approximately reproduces the ‘typical’ distribution shape in Fig. 1C.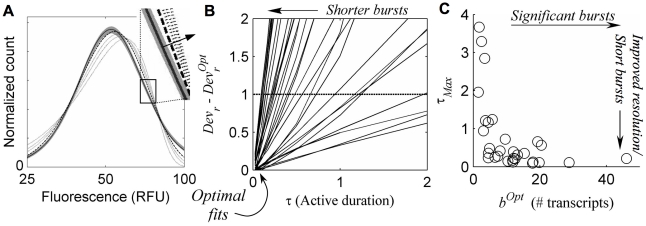
 (short dash), demonstrating increased deviations (τ increasing along arrow in for inset). B) Relative fit deviation,
Devr
decreases for shorter active duration τ, for each clone (solid lines), and are optimal (Devr = DevrOpt) when burst durations are shortest (i.e. in the bursting regime). Devr−DevrOpt = 1 (dashed line) is considered a cut-off, beyond which fit quality is significantly worse than the optimum, specifying a distinguishability cut-off upper-bound on τ, marked by the intersection of dashed line and the solid lines and referred to as
(short dash), demonstrating increased deviations (τ increasing along arrow in for inset). B) Relative fit deviation,
Devr
decreases for shorter active duration τ, for each clone (solid lines), and are optimal (Devr = DevrOpt) when burst durations are shortest (i.e. in the bursting regime). Devr−DevrOpt = 1 (dashed line) is considered a cut-off, beyond which fit quality is significantly worse than the optimum, specifying a distinguishability cut-off upper-bound on τ, marked by the intersection of dashed line and the solid lines and referred to as  for each clone. C) Resolution of bursting dynamics: Calculated upper-bound active-duration (
for each clone. C) Resolution of bursting dynamics: Calculated upper-bound active-duration ( ) and optimal transcriptional burst size (bOpt) for each clone. Predicted large transcriptional bursts (bOpt≫1) identify clones for which the inferred transcriptional dynamics differ significantly from continuous transcription at a single fixed rate, and small
) and optimal transcriptional burst size (bOpt) for each clone. Predicted large transcriptional bursts (bOpt≫1) identify clones for which the inferred transcriptional dynamics differ significantly from continuous transcription at a single fixed rate, and small  indicates good resolution of short bursts from less ‘noisy’ dynamics.
indicates good resolution of short bursts from less ‘noisy’ dynamics.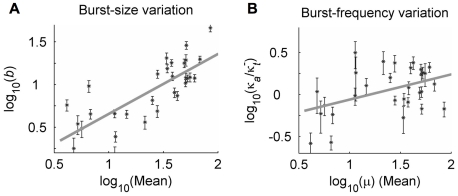
 (which specifies a short active duration that was nearly optimal for all clones). Error bars represent the maximum 95% confidence interval for simultaneous parameter variations that increase Devr by 1. Log-log regression coefficients represent power-law scaling of fit parameters with distribution mean (μ, measured in cytometer RFU), of the form
(which specifies a short active duration that was nearly optimal for all clones). Error bars represent the maximum 95% confidence interval for simultaneous parameter variations that increase Devr by 1. Log-log regression coefficients represent power-law scaling of fit parameters with distribution mean (μ, measured in cytometer RFU), of the form  (x = b or κa), and are given with 95% confidence intervals. A) b: α = 0.76±0.14; β = 0.13±0.2; R2 = 0.66. B) κa: α = 0.2±0.15; β = −0.5±0.2; R2 = 0.2.
(x = b or κa), and are given with 95% confidence intervals. A) b: α = 0.76±0.14; β = 0.13±0.2; R2 = 0.66. B) κa: α = 0.2±0.15; β = −0.5±0.2; R2 = 0.2.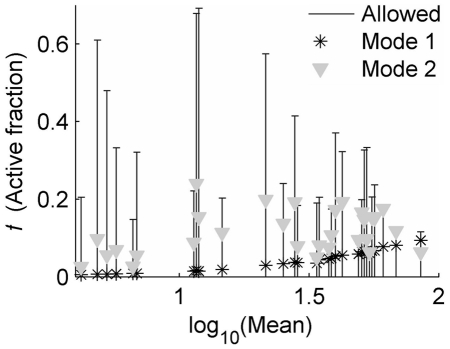
 ), which is marked by bars for each clone. Each Mode of integration-site modulation of transcriptional dynamics leads to a different expected variation of
), which is marked by bars for each clone. Each Mode of integration-site modulation of transcriptional dynamics leads to a different expected variation of  that distinguishes them. For Mode 1, active-state stability varies over integration clones, with the active-state transcription rate fixed (
that distinguishes them. For Mode 1, active-state stability varies over integration clones, with the active-state transcription rate fixed ( was used for this example), while for Mode 2 the active-state transcription rate varies over integrations, with the active duration fixed (
was used for this example), while for Mode 2 the active-state transcription rate varies over integrations, with the active duration fixed ( was used for this example).
was used for this example).
Similar articles
-
Transcriptional bursting from the HIV-1 promoter is a significant source of stochastic noise in HIV-1 gene expression.Biophys J. 2010 Apr 21;98(8):L32-4. doi: 10.1016/j.bpj.2010.03.001. Biophys J. 2010. PMID: 20409455 Free PMC article.
-
HIV Provirus Stably Reproduces Parental Latent and Induced Transcription Phenotypes Regardless of the Chromosomal Integration Site.J Virol. 2016 May 12;90(11):5302-14. doi: 10.1128/JVI.02842-15. Print 2016 Jun 1. J Virol. 2016. PMID: 26984732 Free PMC article.
-
Promoter trapping reveals significant differences in integration site selection between MLV and HIV vectors in primary hematopoietic cells.Blood. 2005 Mar 15;105(6):2307-15. doi: 10.1182/blood-2004-03-0798. Epub 2004 Nov 12. Blood. 2005. PMID: 15542582
-
The Clonal Expansion Dynamics of the HIV-1 Reservoir: Mechanisms of Integration Site-Dependent Proliferation and HIV-1 Persistence.Viruses. 2021 Sep 17;13(9):1858. doi: 10.3390/v13091858. Viruses. 2021. PMID: 34578439 Free PMC article. Review.
-
The Multifaceted Contributions of Chromatin to HIV-1 Integration, Transcription, and Latency.Int Rev Cell Mol Biol. 2017;328:197-252. doi: 10.1016/bs.ircmb.2016.08.006. Epub 2016 Oct 5. Int Rev Cell Mol Biol. 2017. PMID: 28069134 Review.
Cited by
-
Transcriptional Bursting Explains the Noise-Versus-Mean Relationship in mRNA and Protein Levels.PLoS One. 2016 Jul 28;11(7):e0158298. doi: 10.1371/journal.pone.0158298. eCollection 2016. PLoS One. 2016. PMID: 27467384 Free PMC article.
-
Quantifying the contribution of chromatin dynamics to stochastic gene expression reveals long, locus-dependent periods between transcriptional bursts.BMC Biol. 2013 Feb 25;11:15. doi: 10.1186/1741-7007-11-15. BMC Biol. 2013. PMID: 23442824 Free PMC article.
-
Chromatin accessibility at the HIV LTR promoter sets a threshold for NF-κB mediated viral gene expression.Integr Biol (Camb). 2012 Jun;4(6):661-71. doi: 10.1039/c2ib20009k. Epub 2012 May 3. Integr Biol (Camb). 2012. PMID: 22555315 Free PMC article.
-
Gene transcription in bursting: a unified mode for realizing accuracy and stochasticity.Biol Rev Camb Philos Soc. 2019 Feb;94(1):248-258. doi: 10.1111/brv.12452. Epub 2018 Jul 19. Biol Rev Camb Philos Soc. 2019. PMID: 30024089 Free PMC article.
-
Assessing sufficiency and necessity of enhancer activities for gene expression and the mechanisms of transcription activation.Genes Dev. 2018 Feb 1;32(3-4):202-223. doi: 10.1101/gad.310367.117. Genes Dev. 2018. PMID: 29491135 Free PMC article. Review.
References
-
- Tuckwell HC, Shipman PD, Perelson AS. The probability of HIV infection in a new host and its reduction with microbicides. Math Biosci. 2008;214:81–86. - PubMed
-
- Weinberger LS, Burnett JC, Toettcher JE, Arkin AP, Schaffer DV. Stochastic gene expression in a lentiviral positive-feedback loop: HIV-1 Tat fluctuations drive phenotypic diversity. Cell. 2005;122:169–182. - PubMed
-
- Marks AJ, Pillay D, McLean AR. The effect of intrinsic stochasticity on transmitted HIV drug resistance patterns. J Theor Biol. 2009;262:1–13. - PubMed
