

Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis
- PMID: 20941385
- PMCID: PMC2947979
- DOI: 10.1371/journal.pgen.1001141
Genome-wide double-stranded RNA sequencing reveals the functional significance of base-paired RNAs in Arabidopsis
Abstract
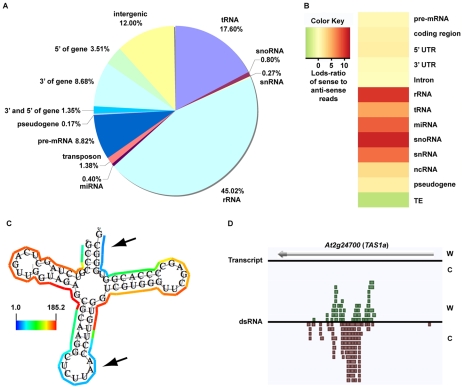
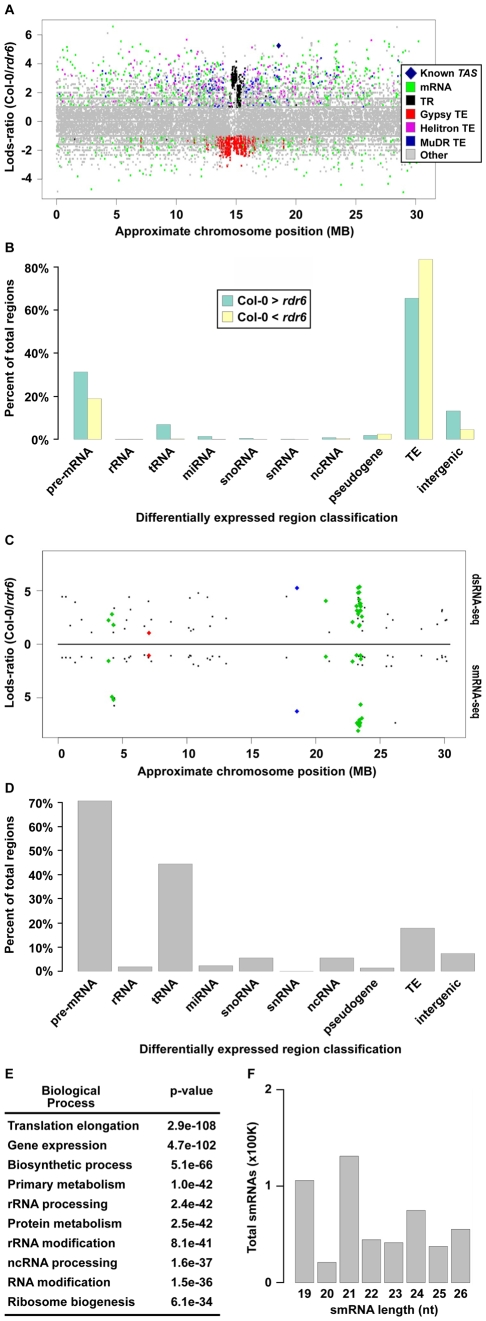
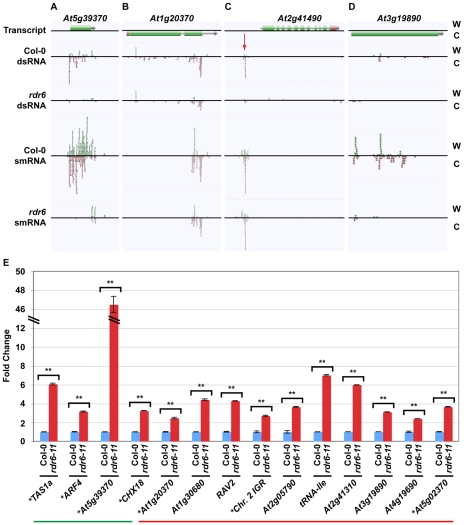
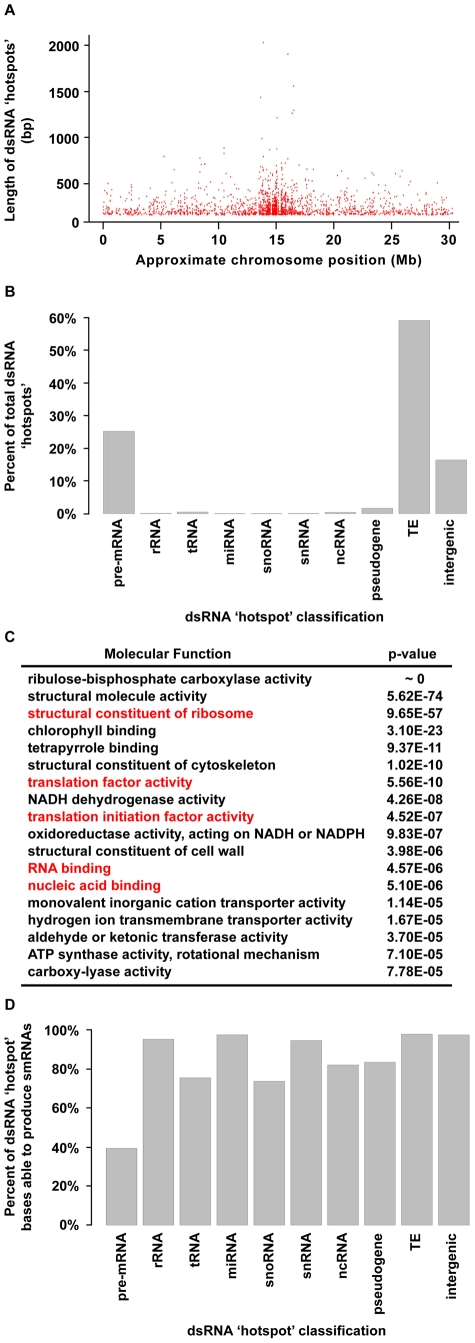
The functional structure of all biologically active molecules is dependent on intra- and inter-molecular interactions. This is especially evident for RNA molecules whose functionality, maturation, and regulation require formation of correct secondary structure through encoded base-pairing interactions. Unfortunately, intra- and inter-molecular base-pairing information is lacking for most RNAs. Here, we marry classical nuclease-based structure mapping techniques with high-throughput sequencing technology to interrogate all base-paired RNA in Arabidopsis thaliana and identify ∼200 new small (sm)RNA-producing substrates of RNA-DEPENDENT RNA POLYMERASE6. Our comprehensive analysis of paired RNAs reveals conserved functionality within introns and both 5' and 3' untranslated regions (UTRs) of mRNAs, as well as a novel population of functional RNAs, many of which are the precursors of smRNAs. Finally, we identify intra-molecular base-pairing interactions to produce a genome-wide collection of RNA secondary structure models. Although our methodology reveals the pairing status of RNA molecules in the absence of cellular proteins, previous studies have demonstrated that structural information obtained for RNAs in solution accurately reflects their structure in ribonucleoprotein complexes. Furthermore, our identification of RNA-DEPENDENT RNA POLYMERASE6 substrates and conserved functional RNA domains within introns and both 5' and 3' untranslated regions (UTRs) of mRNAs using this approach strongly suggests that RNA molecules are correctly folded into their secondary structure in solution. Overall, our findings highlight the importance of base-paired RNAs in eukaryotes and present an approach that should be widely applicable for the analysis of this key structural feature of RNA.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
In vivo genome-wide profiling of RNA secondary structure reveals novel regulatory features.Nature. 2014 Jan 30;505(7485):696-700. doi: 10.1038/nature12756. Epub 2013 Nov 24. Nature. 2014. PMID: 24270811
-
Regulatory impact of RNA secondary structure across the Arabidopsis transcriptome.Plant Cell. 2012 Nov;24(11):4346-59. doi: 10.1105/tpc.112.104232. Epub 2012 Nov 13. Plant Cell. 2012. PMID: 23150631 Free PMC article.
-
Genome-wide probing RNA structure with the modified DMS-MaPseq in Arabidopsis.Methods. 2019 Feb 15;155:30-40. doi: 10.1016/j.ymeth.2018.11.018. Epub 2018 Nov 29. Methods. 2019. PMID: 30503825
-
UTR-Dependent Control of Gene Expression in Plants.Trends Plant Sci. 2018 Mar;23(3):248-259. doi: 10.1016/j.tplants.2017.11.003. Epub 2017 Dec 6. Trends Plant Sci. 2018. PMID: 29223924 Free PMC article. Review.
-
The Conservation and Function of RNA Secondary Structure in Plants.Annu Rev Plant Biol. 2016 Apr 29;67:463-88. doi: 10.1146/annurev-arplant-043015-111754. Epub 2016 Feb 8. Annu Rev Plant Biol. 2016. PMID: 26865341 Free PMC article. Review.
Cited by
-
The role of the Arabidopsis Exosome in siRNA-independent silencing of heterochromatic loci.PLoS Genet. 2013 Mar;9(3):e1003411. doi: 10.1371/journal.pgen.1003411. Epub 2013 Mar 28. PLoS Genet. 2013. PMID: 23555312 Free PMC article.
-
Bridging the gap between in vitro and in vivo RNA folding.Q Rev Biophys. 2016 Jan;49:e10. doi: 10.1017/S003358351600007X. Epub 2016 Jun 24. Q Rev Biophys. 2016. PMID: 27658939 Free PMC article.
-
Global Analysis of RNA-Dependent RNA Polymerase-Dependent Small RNAs Reveals New Substrates and Functions for These Proteins and SGS3 in Arabidopsis.Noncoding RNA. 2021 Apr 27;7(2):28. doi: 10.3390/ncrna7020028. Noncoding RNA. 2021. PMID: 33925339 Free PMC article.
-
StructureFold: genome-wide RNA secondary structure mapping and reconstruction in vivo.Bioinformatics. 2015 Aug 15;31(16):2668-75. doi: 10.1093/bioinformatics/btv213. Epub 2015 Apr 16. Bioinformatics. 2015. PMID: 25886980 Free PMC article.
-
High-throughput approaches for plant epigenomic studies.Curr Opin Plant Biol. 2011 Apr;14(2):130-6. doi: 10.1016/j.pbi.2011.03.010. Epub 2011 Apr 4. Curr Opin Plant Biol. 2011. PMID: 21470901 Free PMC article. Review.
References
-
- Cruz JA, Westhof E. The dynamic landscapes of RNA architecture. Cell. 2009;136:604–609. - PubMed
-
- Mendell JT, Dietz HC. When the message goes awry: disease-producing mutations that influence mRNA content and performance. Cell. 2001;107:411–414. - PubMed
-
- Montange RK, Batey RT. Riboswitches: emerging themes in RNA structure and function. Annu Rev Biophys. 2008;37:117–133. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
