

Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses
- PMID: 20889916
- PMCID: PMC3476703
- DOI: 10.1158/1078-0432.CCR-10-0712
Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses
Abstract
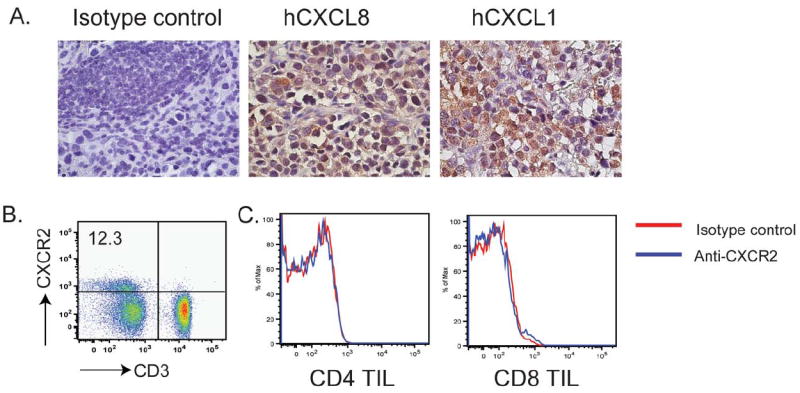
Purpose: One of the most important rate-limiting steps in adoptive cell transfer (ACT) is the inefficient migration of T cells to tumors. Because melanomas specifically express the chemokines CXCL1 and CXCL8 that are known to facilitate the CXCR2-dependent migration by monocytes, our aim is to evaluate whether introduction of the CXCR2 gene into tumor-specific T cells could further improve the effectiveness of ACT by enhancing T-cell migration to tumor.
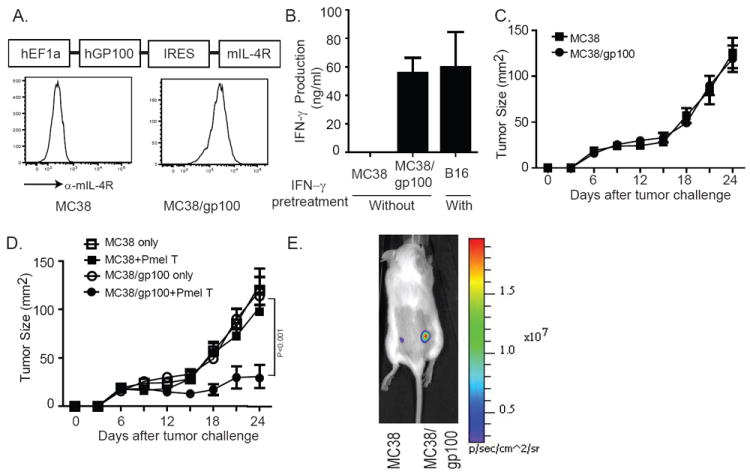
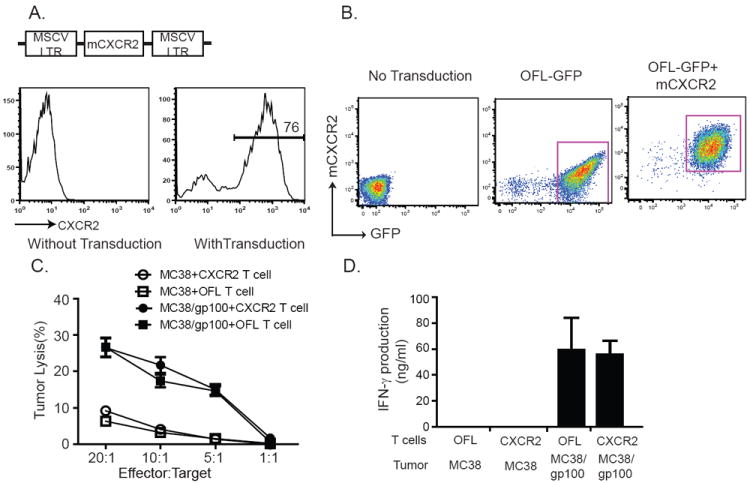
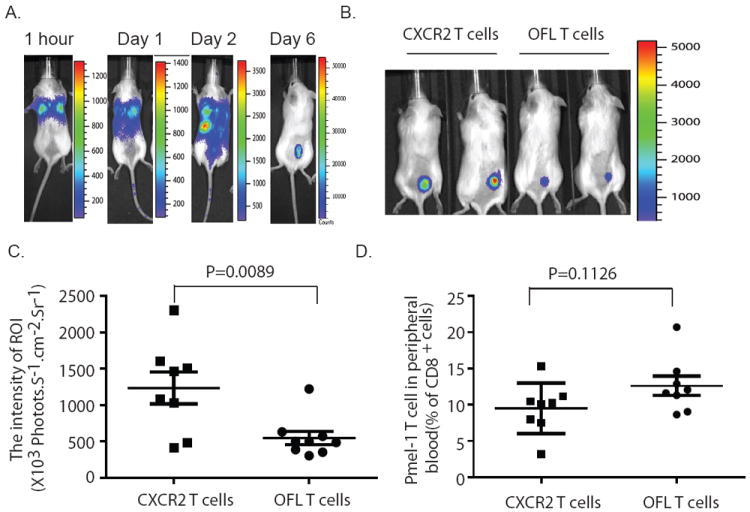
Experimental design: In this study, we used transgenic pmel-1 T cells, which recognize gp100 in the context of H-2Db, that were transduced with luciferase gene to monitor the migration of transferred T cells in vivo. To visualize luciferase-expressing T cells within a tumor, a nonpigmented tumor is required. Therefore, we used the MC38 tumor model, which naturally expresses CXCL1.
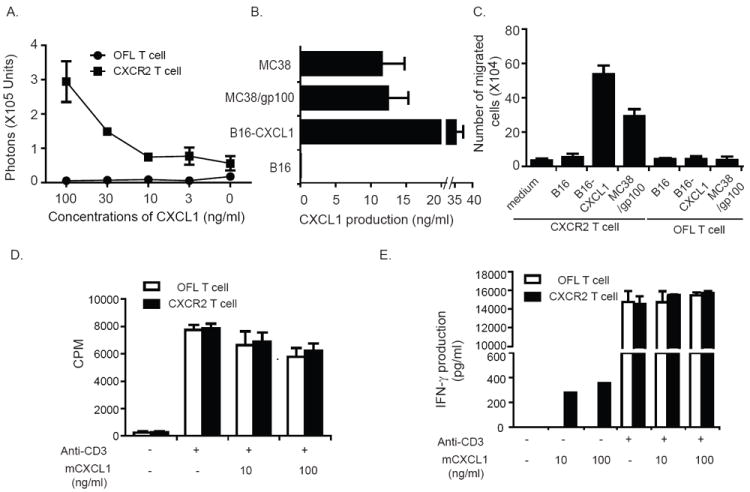
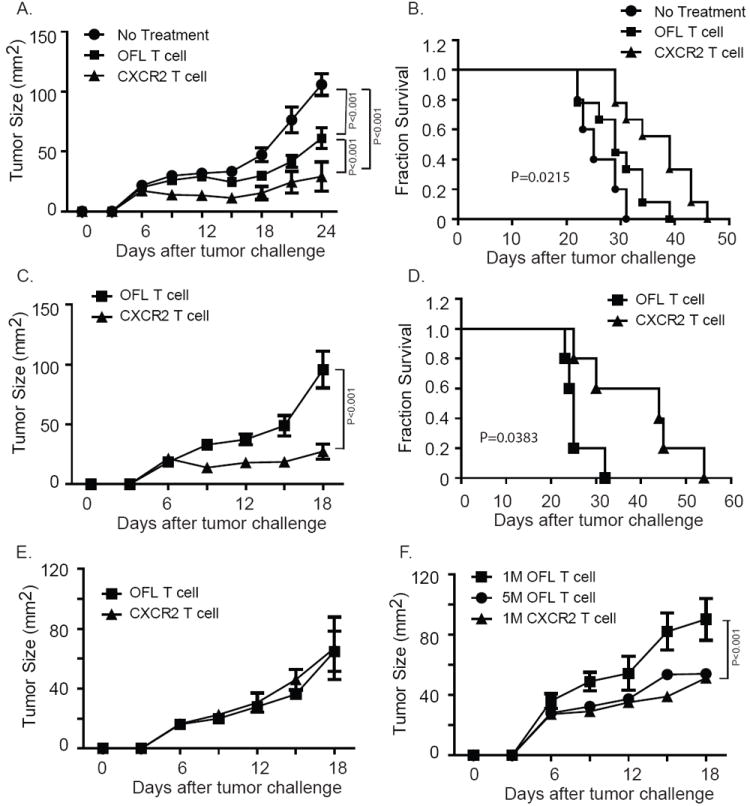
Results: Mice bearing MC38/gp100 tumor cells treated with CXCR2/luciferase-transduced pmel-1 T cells showed enhanced tumor regression and survival compared with mice receiving control luciferase-transduced pmel-1 T cells. We also observed preferential accumulation of CXCR2-expressing pmel-1 T cells in the tumor sites of these mice using bioluminescence imaging. A similar enhancement in tumor regression and survival was observed when CXCR2-transduced pmel-1 T cells were transferred into mice bearing CXCL1-transduced B16 tumors compared with mice treated with control pmel-1 T cells.
Conclusions: These results implicate that the introduction of the CXCR2 gene into tumor-specific T cells can enhance their localization to tumors and improve antitumor immune responses. This strategy may ultimately enable personalization of cancer therapies based on chemokine expression by tumors.
©2010 AACR.
Figures
Similar articles
-
Redirecting migration of T cells to chemokine secreted from tumors by genetic modification with CXCR2.Hum Gene Ther. 2002 Nov 1;13(16):1971-80. doi: 10.1089/10430340260355374. Hum Gene Ther. 2002. PMID: 12427307
-
CXCR2-mediated tumor-associated neutrophil recruitment is regulated by IFN-β.Int J Cancer. 2014 Mar 15;134(6):1346-58. doi: 10.1002/ijc.28551. Epub 2013 Oct 31. Int J Cancer. 2014. PMID: 24154944
-
Genetic engineering of human NK cells to express CXCR2 improves migration to renal cell carcinoma.J Immunother Cancer. 2017 Sep 19;5(1):73. doi: 10.1186/s40425-017-0275-9. J Immunother Cancer. 2017. PMID: 28923105 Free PMC article.
-
T-cell receptor gene therapy of established tumors in a murine melanoma model.J Immunother. 2008 Jan;31(1):1-6. doi: 10.1097/CJI.0b013e31815c193f. J Immunother. 2008. PMID: 18157006 Free PMC article.
-
CXCR2 Receptor: Regulation of Expression, Signal Transduction, and Involvement in Cancer.Int J Mol Sci. 2022 Feb 16;23(4):2168. doi: 10.3390/ijms23042168. Int J Mol Sci. 2022. PMID: 35216283 Free PMC article. Review.
Cited by
-
Adoptive T Cell Therapy for Solid Tumors: Pathway to Personalized Standard of Care.Cells. 2021 Apr 5;10(4):808. doi: 10.3390/cells10040808. Cells. 2021. PMID: 33916369 Free PMC article. Review.
-
Beyond immune checkpoint blockade: emerging immunological strategies.Nat Rev Drug Discov. 2021 Dec;20(12):899-919. doi: 10.1038/s41573-021-00155-y. Epub 2021 Mar 8. Nat Rev Drug Discov. 2021. PMID: 33686237 Review.
-
'Off-the-shelf' allogeneic CAR T cells: development and challenges.Nat Rev Drug Discov. 2020 Mar;19(3):185-199. doi: 10.1038/s41573-019-0051-2. Epub 2020 Jan 3. Nat Rev Drug Discov. 2020. PMID: 31900462 Review.
-
Chemokine receptor engineering of T cells with CXCR2 improves homing towards subcutaneous human melanomas in xenograft mouse model.Oncoimmunology. 2018 Apr 18;7(8):e1450715. doi: 10.1080/2162402X.2018.1450715. eCollection 2018. Oncoimmunology. 2018. PMID: 30221044 Free PMC article.
-
Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment.J Immunother Cancer. 2017 Mar 21;5:28. doi: 10.1186/s40425-017-0230-9. eCollection 2017. J Immunother Cancer. 2017. PMID: 28331617 Free PMC article. Review.
References
-
- Pockaj BA, Sherry RM, Wei JP, et al. Localization of 111indium-labeled tumor infiltrating lymphocytes to tumor in patients receiving adoptive immunotherapy. Augmentation with cyclophosphamide and correlation with response. Cancer. 1994;73:1731–7. - PubMed
-
- Fisher B, Packard BS, Read EJ, et al. Tumor localization of adoptively transferred indium-111 labeled tumor infiltrating lymphocytes in patients with metastatic melanoma. J Clin Oncol. 1989;7:250–61. - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
