

A positive role for c-Abl in Atm and Atr activation in DNA damage response
- PMID: 20798688
- PMCID: PMC3131864
- DOI: 10.1038/cdd.2010.106
A positive role for c-Abl in Atm and Atr activation in DNA damage response
Abstract
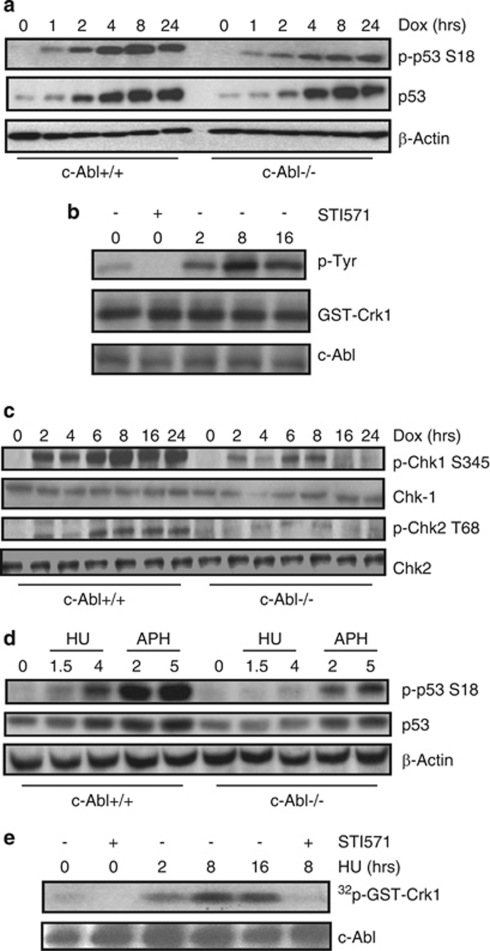
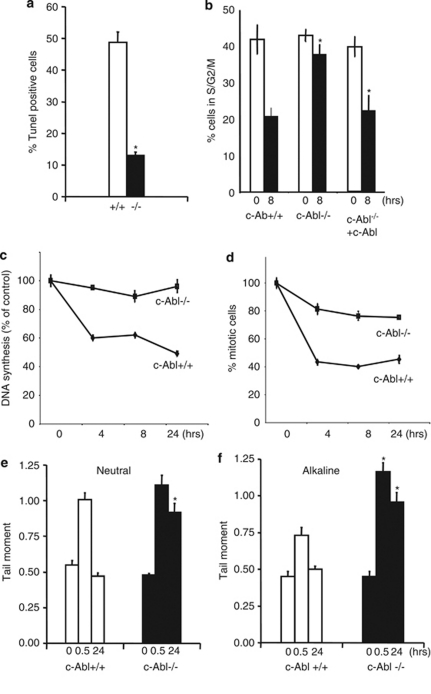
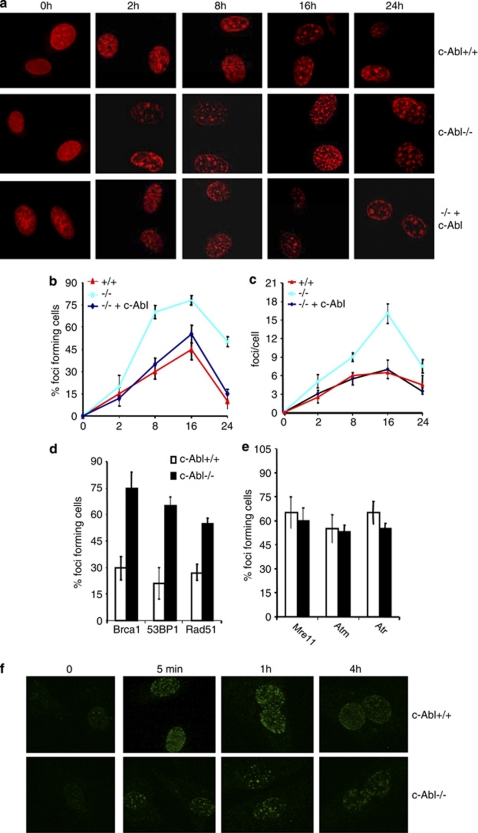
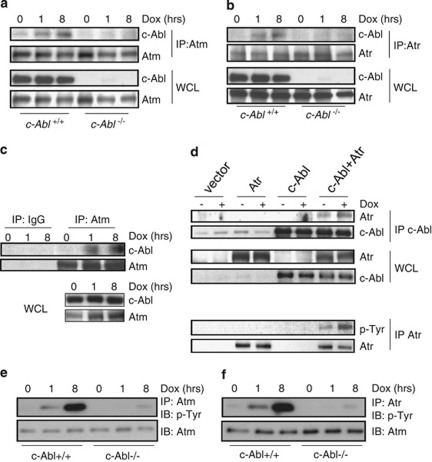
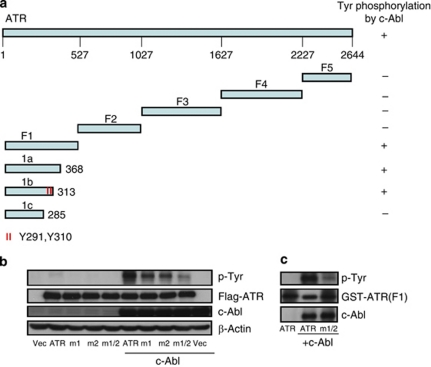
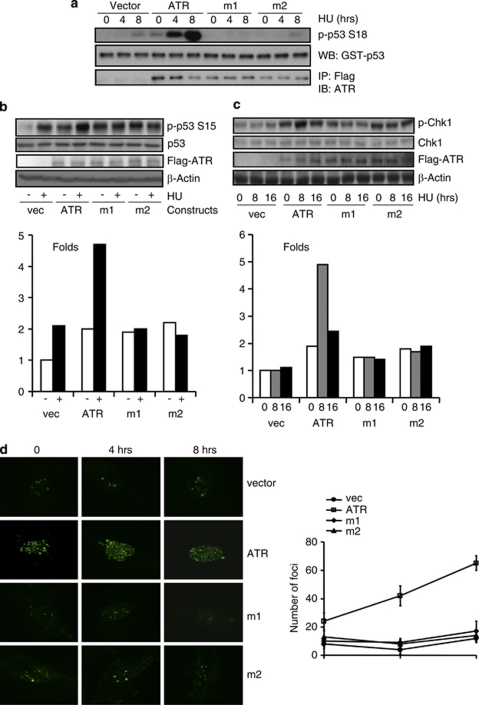
DNA damage triggers Atm- and/or Atr-dependent signaling pathways to control cell cycle progression, apoptosis, and DNA repair. However, how Atm and Atr are activated is not fully understood. One of the downstream targets of Atm is non-receptor tyrosine kinase c-Abl, which is phosphorylated and activated by Atm. The current view is that c-Abl relays pro-apoptotic signals from Atm to p73 and p53. Here we show that c-Abl deficiency resulted in a broad spectrum of defects in cell response to genotoxic stress, including activation of Chk1 and Chk2, activation of p53, nuclear foci formation, apoptosis, and DNA repair, suggesting that c-Abl might also act upstream of the DNA damage-activated signaling cascades in addition to its role in p73 and p53 regulation. Indeed, we found that c-Abl is required for proper activation of both Atm and Atr. c-Abl is bound to the chromatin and shows enhanced interaction with Atm and Atr in response to DNA damage. c-Abl can phosphorylate Atr on Y291 and Y310 and this phosphorylation appears to have a positive role in Atr activation under genotoxic stress. These findings suggest that Atm-mediated c-Abl activation in cell response to double-stranded DNA breaks might facilitate the activation of both Atm and Atr to regulate their downstream cellular events.
Figures

Comment in
-
c-Abl tyrosine kinase in the DNA damage response: cell death and more.Cell Death Differ. 2011 Jan;18(1):2-4. doi: 10.1038/cdd.2010.132. Cell Death Differ. 2011. PMID: 21151157 Free PMC article. No abstract available.
Similar articles
-
ATR-Chk2 signaling in p53 activation and DNA damage response during cisplatin-induced apoptosis.J Biol Chem. 2008 Mar 7;283(10):6572-83. doi: 10.1074/jbc.M707568200. Epub 2007 Dec 27. J Biol Chem. 2008. PMID: 18162465
-
Ataxia telangiectasia mutated (ATM) and ATM and Rad3-related protein exhibit selective target specificities in response to different forms of DNA damage.J Biol Chem. 2005 Jan 14;280(2):1186-92. doi: 10.1074/jbc.M410873200. Epub 2004 Nov 8. J Biol Chem. 2005. PMID: 15533933
-
ATR and Chk1 suppress a caspase-3-dependent apoptotic response following DNA replication stress.PLoS Genet. 2009 Jan;5(1):e1000324. doi: 10.1371/journal.pgen.1000324. Epub 2009 Jan 2. PLoS Genet. 2009. PMID: 19119425 Free PMC article.
-
Determination of cell fate by c-Abl activation in the response to DNA damage.Oncogene. 1998 Dec 24;17(25):3309-18. doi: 10.1038/sj.onc.1202571. Oncogene. 1998. PMID: 9916993 Review.
-
The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer.Adv Cancer Res. 2010;108:73-112. doi: 10.1016/B978-0-12-380888-2.00003-0. Adv Cancer Res. 2010. PMID: 21034966 Review.
Cited by
-
The Tyrosine Kinase c-Abl Promotes Homeodomain-interacting Protein Kinase 2 (HIPK2) Accumulation and Activation in Response to DNA Damage.J Biol Chem. 2015 Jul 3;290(27):16478-88. doi: 10.1074/jbc.M114.628982. Epub 2015 May 5. J Biol Chem. 2015. PMID: 25944899 Free PMC article.
-
Comparative Phosphoproteomic Analysis of Sporulated Oocysts and Tachyzoites of Toxoplasma gondii Reveals Stage-Specific Patterns.Molecules. 2022 Feb 2;27(3):1022. doi: 10.3390/molecules27031022. Molecules. 2022. PMID: 35164288 Free PMC article.
-
The p53 family member p73 in the regulation of cell stress response.Biol Direct. 2021 Nov 8;16(1):23. doi: 10.1186/s13062-021-00307-5. Biol Direct. 2021. PMID: 34749806 Free PMC article. Review.
-
C2orf40 inhibits metastasis and regulates chemo-resistance and radio-resistance of nasopharyngeal carcinoma cells by influencing cell cycle and activating the PI3K/AKT/mTOR signaling pathway.J Transl Med. 2022 Jun 8;20(1):264. doi: 10.1186/s12967-022-03446-z. J Transl Med. 2022. PMID: 35676661 Free PMC article.
-
Rho GTPases: Novel Players in the Regulation of the DNA Damage Response?Biomolecules. 2015 Sep 30;5(4):2417-34. doi: 10.3390/biom5042417. Biomolecules. 2015. PMID: 26437439 Free PMC article. Review.
References
-
- Peterson CL, Cote J. Cellular machineries for chromosomal DNA repair. Genes Dev. 2004;18:602–616. - PubMed
-
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev. 2001;15:2177–2196. - PubMed
-
- Shiloh Y. ATM and related protein kinases: safeguarding genome integrity. Nat Rev Cancer. 2003;3:155–168. - PubMed
-
- Cline SD, Hanawalt PC. Who's on first in the cellular response to DNA damage. Nat Rev Mol Cell Biol. 2003;4:361–372. - PubMed
-
- D'Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol. 2002;3:317–327. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
