

Hypoxia-inducible factor 1alpha determines gastric cancer chemosensitivity via modulation of p53 and NF-kappaB
- PMID: 20706634
- PMCID: PMC2919384
- DOI: 10.1371/journal.pone.0012038
Hypoxia-inducible factor 1alpha determines gastric cancer chemosensitivity via modulation of p53 and NF-kappaB
Abstract
Background: Reduced chemosensitivity of solid cancer cells represents a pivotal obstacle in clinical oncology. Hence, the molecular characterization of pathways regulating chemosensitivity is a central prerequisite to improve cancer therapy. The hypoxia-inducible factor HIF-1alpha has been linked to chemosensitivity while the underlying molecular mechanisms remain largely elusive. Therefore, we comprehensively analysed HIF-1alpha's role in determining chemosensitivity focussing on responsible molecular pathways.
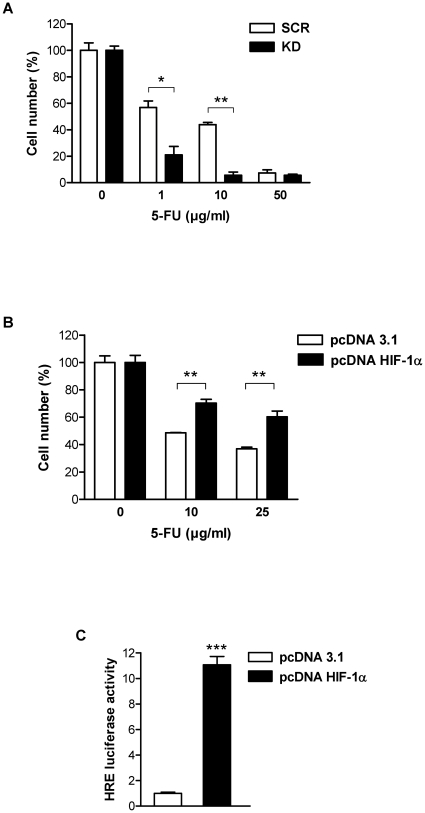
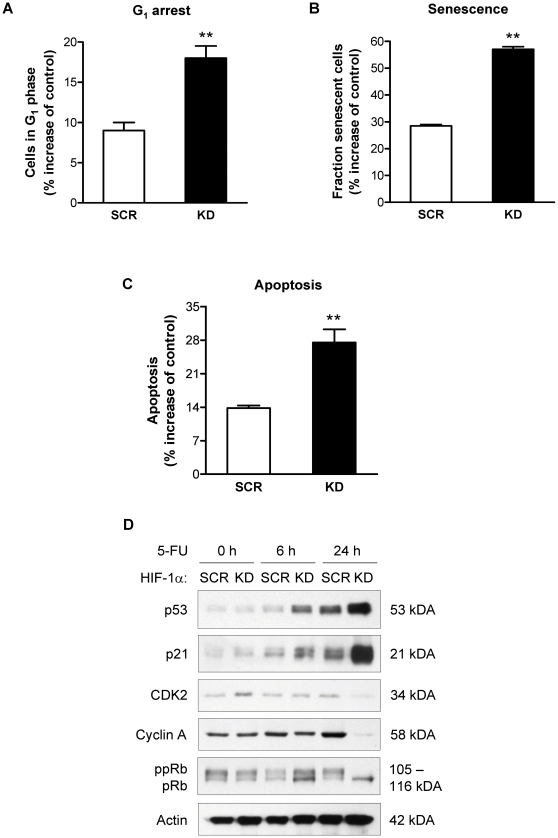
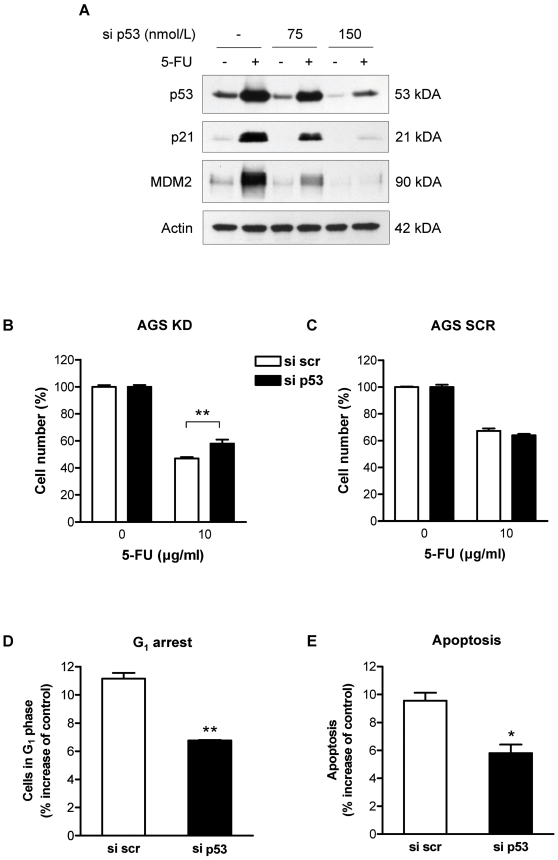
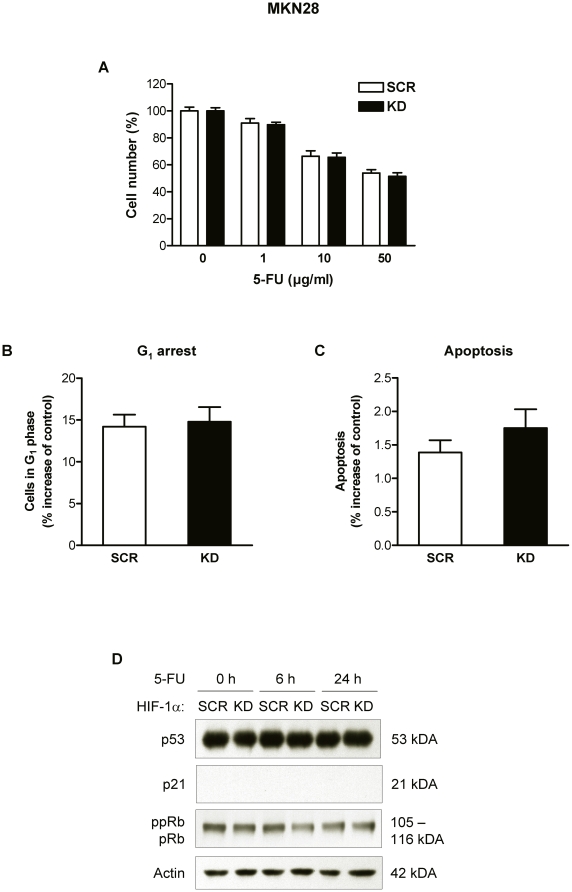
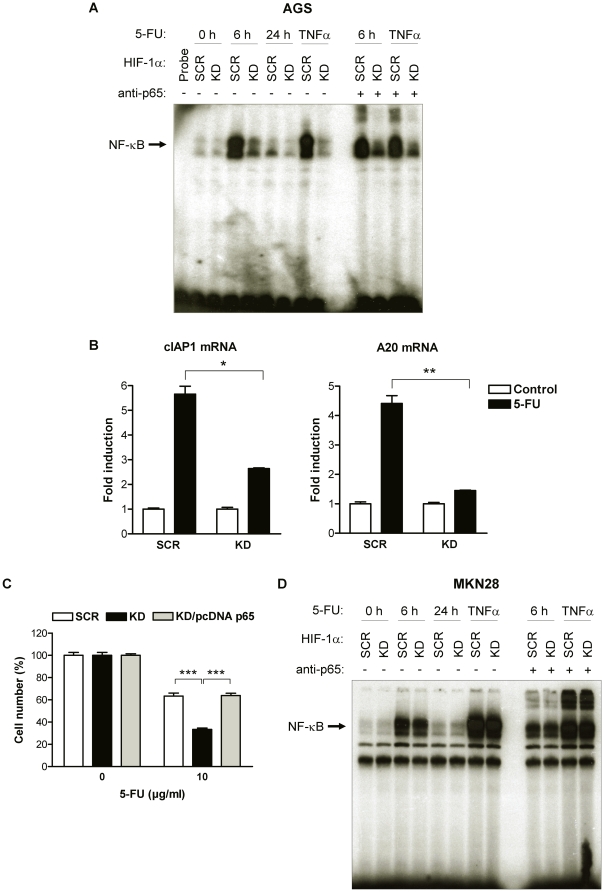
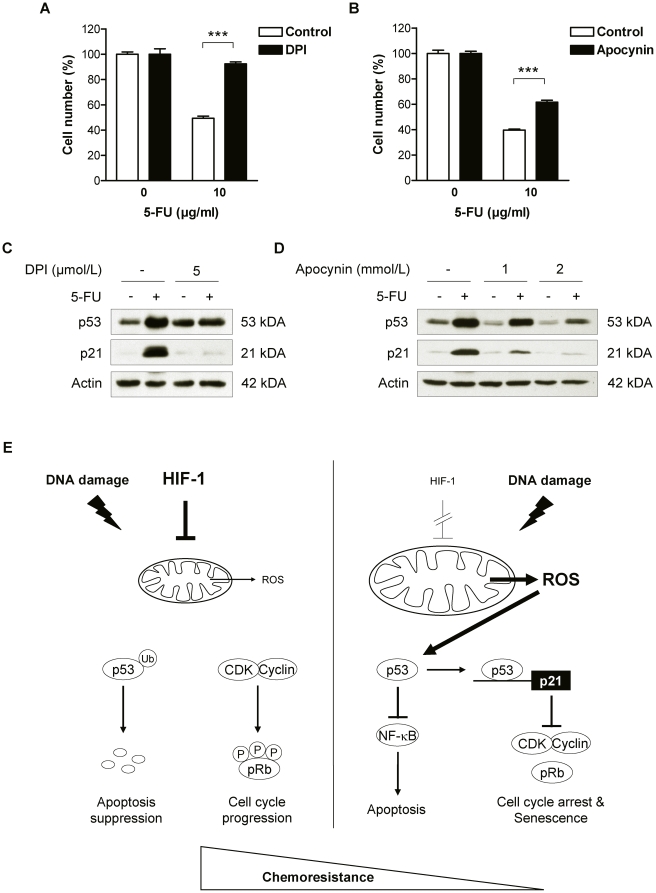
Methodology and principal findings: RNA interference was applied to inactivate HIF-1alpha or p53 in the human gastric cancer cell lines AGS and MKN28. The chemotherapeutic agents 5-fluorouracil and cisplatin were used and chemosensitivity was assessed by cell proliferation assays as well as determination of cell cycle distribution and apoptosis. Expression of p53 and p53 target proteins was analyzed by western blot. NF-kappaB activity was characterized by means of electrophoretic mobility shift assay. Inactivation of HIF-1alpha in gastric cancer cells resulted in robust elevation of chemosensitivity. Accordingly, HIF-1alpha-competent cells displayed a significant reduction of chemotherapy-induced senescence and apoptosis. Remarkably, this phenotype was completely absent in p53 mutant cells while inactivation of p53 per se did not affect chemosensitivity. HIF-1alpha markedly suppressed chemotherapy-induced activation of p53 and p21 as well as the retinoblastoma protein, eventually resulting in cell cycle arrest. Reduced formation of reactive oxygen species in HIF-1alpha-competent cells was identified as the molecular mechanism of HIF-1alpha-mediated inhibition of p53. Furthermore, loss of HIF-1alpha abrogated, in a p53-dependent manner, chemotherapy-induced DNA-binding of NF-kappaB and expression of anti-apoptotic NF-kappaB target genes. Accordingly, reconstitution of the NF-kappaB subunit p65 reversed the increased chemosensitivity of HIF-1alpha-deficient cells.
Conclusion and significance: In summary, we identified HIF-1alpha as a potent regulator of p53 and NF-kappaB activity under conditions of genotoxic stress. We conclude that p53 mutations in human tumors hold the potential to confound the efficacy of HIF-1-inhibitors in cancer therapy.
Conflict of interest statement
Figures
Similar articles
-
A compensatory role of NF-κB to p53 in response to 5-FU-based chemotherapy for gastric cancer cell lines.PLoS One. 2014 Feb 27;9(2):e90155. doi: 10.1371/journal.pone.0090155. eCollection 2014. PLoS One. 2014. PMID: 24587255 Free PMC article.
-
A hypoxia-dependent upregulation of hypoxia-inducible factor-1 by nuclear factor-κB promotes gastric tumour growth and angiogenesis.Br J Cancer. 2011 Jan 4;104(1):166-74. doi: 10.1038/sj.bjc.6606020. Epub 2010 Nov 30. Br J Cancer. 2011. PMID: 21119667 Free PMC article.
-
Effects of lentivirus-mediated HIF-1alpha knockdown on hypoxia-related cisplatin resistance and their dependence on p53 status in fibrosarcoma cells.Cancer Gene Ther. 2008 Jul;15(7):449-55. doi: 10.1038/cgt.2008.4. Epub 2008 Apr 18. Cancer Gene Ther. 2008. PMID: 18421307
-
Hypoxia-mediated drug resistance: novel insights on the functional interaction of HIFs and cell death pathways.Drug Resist Updat. 2011 Jun;14(3):191-201. doi: 10.1016/j.drup.2011.03.001. Epub 2011 Apr 3. Drug Resist Updat. 2011. PMID: 21466972 Review.
-
Is the hypoxia-inducible factor pathway important in gastric cancer?Eur J Cancer. 2005 Dec;41(18):2792-805. doi: 10.1016/j.ejca.2005.09.008. Epub 2005 Nov 14. Eur J Cancer. 2005. PMID: 16290133 Review.
Cited by
-
ER stress in retinal degeneration in S334ter Rho rats.PLoS One. 2012;7(3):e33266. doi: 10.1371/journal.pone.0033266. Epub 2012 Mar 14. PLoS One. 2012. PMID: 22432009 Free PMC article.
-
A Hypoxia Molecular Signature-Based Prognostic Model for Endometrial Cancer Patients.Int J Mol Sci. 2023 Jan 14;24(2):1675. doi: 10.3390/ijms24021675. Int J Mol Sci. 2023. PMID: 36675190 Free PMC article.
-
The Tumor Microenvironment in Tumorigenesis and Therapy Resistance Revisited.Cancers (Basel). 2023 Jan 6;15(2):376. doi: 10.3390/cancers15020376. Cancers (Basel). 2023. PMID: 36672326 Free PMC article. Review.
-
Emerging Nano-Based Strategies Against Drug Resistance in Tumor Chemotherapy.Front Bioeng Biotechnol. 2021 Dec 7;9:798882. doi: 10.3389/fbioe.2021.798882. eCollection 2021. Front Bioeng Biotechnol. 2021. PMID: 34950650 Free PMC article. Review.
-
Hypoxia-inducible factors: mediators of cancer progression; prognostic and therapeutic targets in soft tissue sarcomas.Cancers (Basel). 2013 Apr 2;5(2):320-33. doi: 10.3390/cancers5020320. Cancers (Basel). 2013. PMID: 24216979 Free PMC article.
References
-
- Gatti L, Zunino F. Overview of tumor cell chemoresistance mechanisms. Methods Mol Med. 2005;111:127–148. - PubMed
-
- Gottesman MM. Mechanisms of cancer drug resistance. Annu Rev Med. 2002;53:615–627. - PubMed
-
- Morin PJ. Drug resistance and the microenvironment: nature and nurture. Drug Resist Updat. 2003;6:169–172. - PubMed
-
- Chabner BA, Roberts TG. Chemotherapy and the war on cancer. Nat Rev Cancer. 2005;5:65–72. - PubMed
-
- Wilson TR, Longley DB, Johnston PG. Chemoresistance in solid tumours. Ann Oncol. 2006;17:315–324. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
