

Murine gamma-herpesvirus 68 hijacks MAVS and IKKbeta to initiate lytic replication
- PMID: 20686657
- PMCID: PMC2912392
- DOI: 10.1371/journal.ppat.1001001
Murine gamma-herpesvirus 68 hijacks MAVS and IKKbeta to initiate lytic replication
Abstract
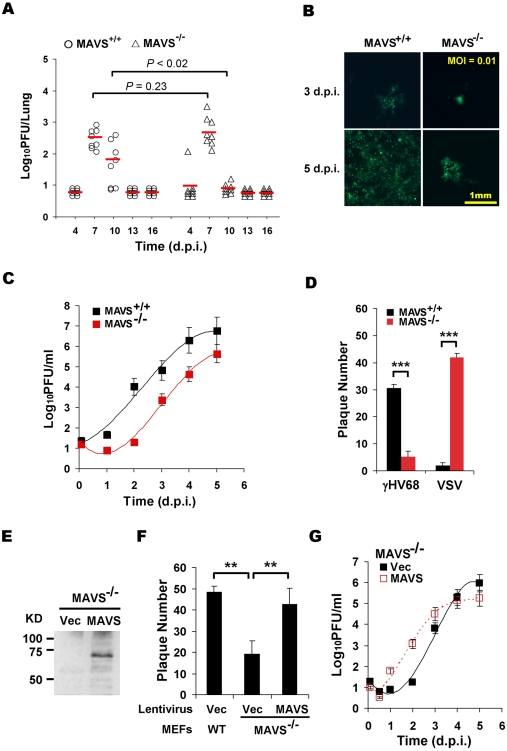
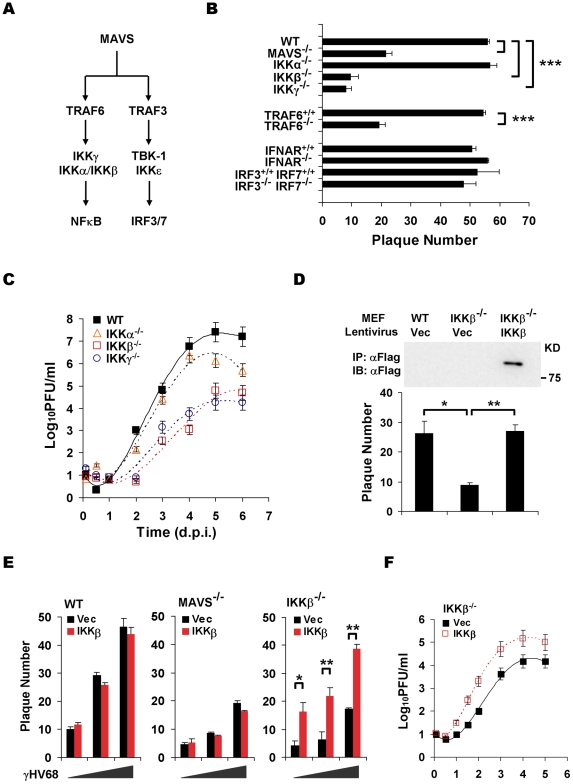
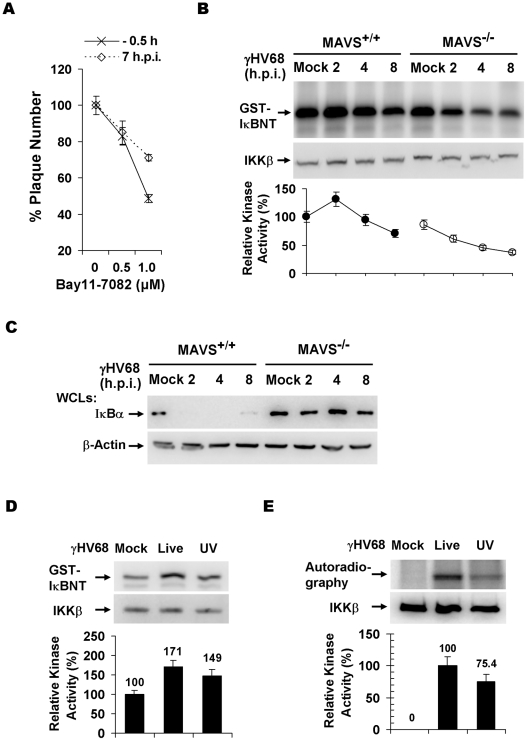
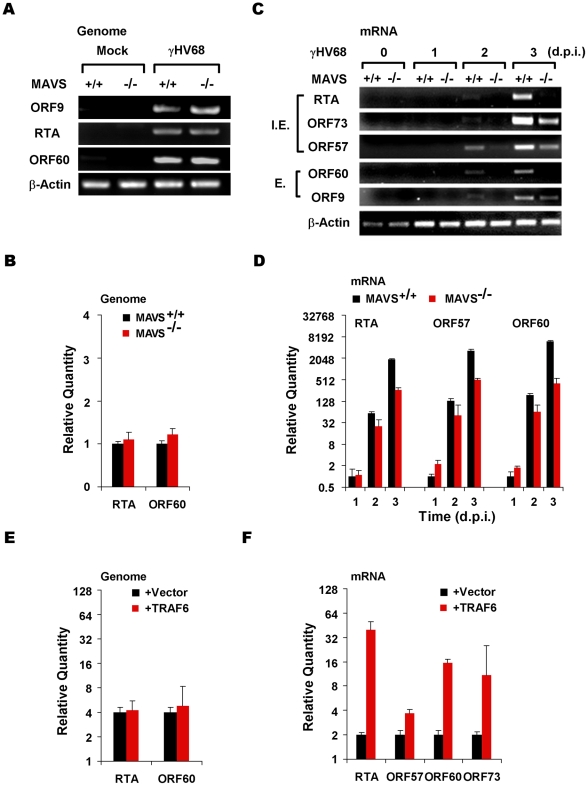
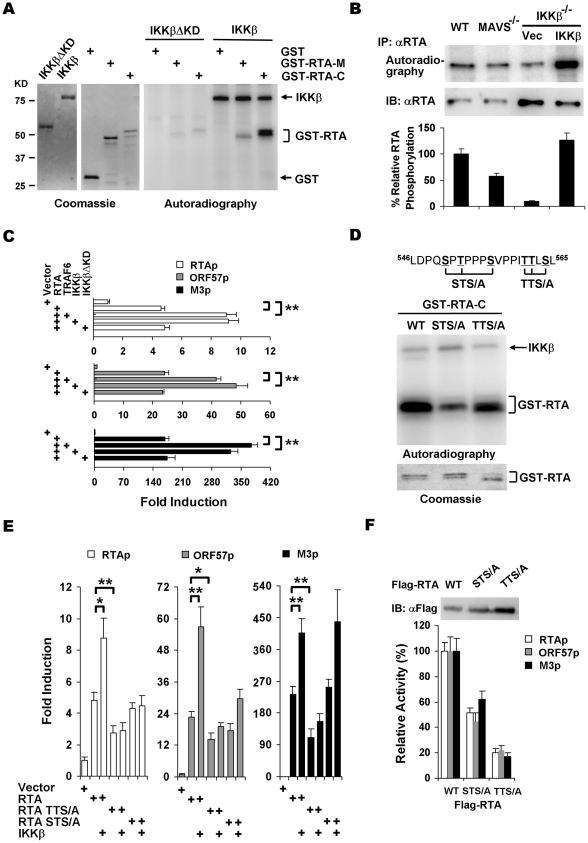
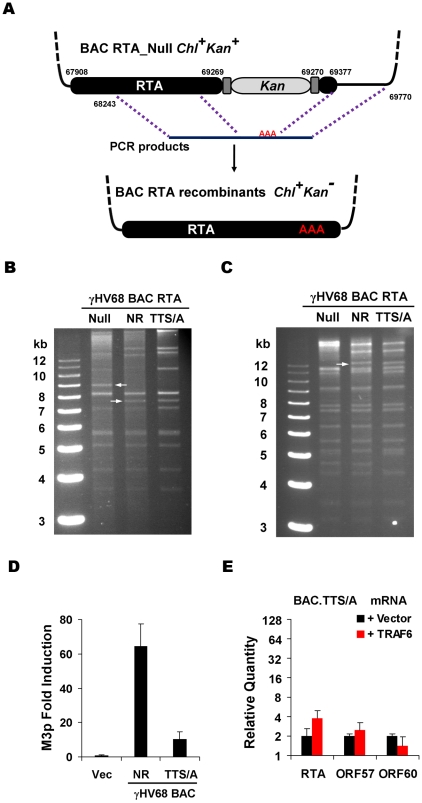
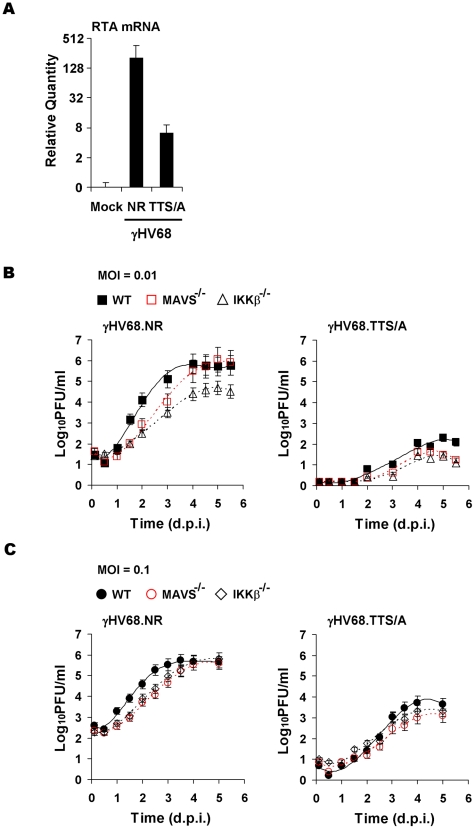
Upon viral infection, the mitochondrial antiviral signaling (MAVS)-IKKbeta pathway is activated to restrict viral replication. Manipulation of immune signaling events by pathogens has been an outstanding theme of host-pathogen interaction. Here we report that the loss of MAVS or IKKbeta impaired the lytic replication of gamma-herpesvirus 68 (gammaHV68), a model herpesvirus for human Kaposi's sarcoma-associated herpesvirus and Epstein-Barr virus. gammaHV68 infection activated IKKbeta in a MAVS-dependent manner; however, IKKbeta phosphorylated and promoted the transcriptional activation of the gammaHV68 replication and transcription activator (RTA). Mutational analyses identified IKKbeta phosphorylation sites, through which RTA-mediated transcription was increased by IKKbeta, within the transactivation domain of RTA. Moreover, the lytic replication of recombinant gammaHV68 carrying mutations within the IKKbeta phosphorylation sites was greatly impaired. These findings support the conclusion that gammaHV68 hijacks the antiviral MAVS-IKKbeta pathway to promote viral transcription and lytic infection, representing an example whereby viral replication is coupled to host immune activation.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
Similar articles
-
Murine gamma herpesvirus 68 hijacks MAVS and IKKβ to abrogate NFκB activation and antiviral cytokine production.PLoS Pathog. 2011 Nov;7(11):e1002336. doi: 10.1371/journal.ppat.1002336. Epub 2011 Nov 10. PLoS Pathog. 2011. PMID: 22110409 Free PMC article.
-
Dissecting host-virus interaction in lytic replication of a model herpesvirus.J Vis Exp. 2011 Oct 7;(56):3140. doi: 10.3791/3140. J Vis Exp. 2011. PMID: 22006158 Free PMC article.
-
Recombinant Murine Gamma Herpesvirus 68 Carrying KSHV G Protein-Coupled Receptor Induces Angiogenic Lesions in Mice.PLoS Pathog. 2015 Jun 24;11(6):e1005001. doi: 10.1371/journal.ppat.1005001. eCollection 2015 Jun. PLoS Pathog. 2015. PMID: 26107716 Free PMC article.
-
The Rta/Orf50 transactivator proteins of the gamma-herpesviridae.Curr Top Microbiol Immunol. 2007;312:71-100. doi: 10.1007/978-3-540-34344-8_3. Curr Top Microbiol Immunol. 2007. PMID: 17089794 Review.
-
Lytic cycle switches of oncogenic human gammaherpesviruses.Adv Cancer Res. 2007;97:81-109. doi: 10.1016/S0065-230X(06)97004-3. Adv Cancer Res. 2007. PMID: 17419942 Review.
Cited by
-
Herpesviral G protein-coupled receptors activate NFAT to induce tumor formation via inhibiting the SERCA calcium ATPase.PLoS Pathog. 2015 Mar 26;11(3):e1004768. doi: 10.1371/journal.ppat.1004768. eCollection 2015 Mar. PLoS Pathog. 2015. PMID: 25811856 Free PMC article.
-
Host Noncoding Retrotransposons Induced by DNA Viruses: a SINE of Infection?J Virol. 2017 Nov 14;91(23):e00982-17. doi: 10.1128/JVI.00982-17. Print 2017 Dec 1. J Virol. 2017. PMID: 28931686 Free PMC article. Review.
-
An Internally Translated MAVS Variant Exposes Its Amino-terminal TRAF-Binding Motifs to Deregulate Interferon Induction.PLoS Pathog. 2015 Jul 29;11(7):e1005060. doi: 10.1371/journal.ppat.1005060. eCollection 2015 Jul. PLoS Pathog. 2015. PMID: 26221961 Free PMC article.
-
Endoplasmic reticulum stress causes EBV lytic replication.Blood. 2011 Nov 17;118(20):5528-39. doi: 10.1182/blood-2011-04-347112. Epub 2011 Aug 17. Blood. 2011. PMID: 21849482 Free PMC article.
-
Alteration of the Premature tRNA Landscape by Gammaherpesvirus Infection.mBio. 2020 Dec 15;11(6):e02664-20. doi: 10.1128/mBio.02664-20. mBio. 2020. PMID: 33323507 Free PMC article.
References
-
- Akira S, Uematsu S, Takeuchi O. Pathogen recognition and innate immunity. Cell. 2006;124:783–801. - PubMed
-
- Medzhitov R. Recognition of microorganisms and activation of the immune response. Nature. 2007;449:819–826. - PubMed
-
- Kawai T, Takahashi K, Sato S, Coban C, Kumar H, et al. IPS-1, an adaptor triggering RIG-I- and Mda5-mediated type I interferon induction. Nat Immunol. 2005;6:981–988. - PubMed
-
- Seth RB, Sun L, Ea CK, Chen ZJ. Identification and characterization of MAVS, a mitochondrial antiviral signaling protein that activates NF-kappaB and IRF 3. Cell. 2005;122:669–682. - PubMed
-
- Xu LG, Wang YY, Han KJ, Li LY, Zhai Z, et al. VISA is an adapter protein required for virus-triggered IFN-beta signaling. Mol Cell. 2005;19:727–740. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
