

Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations
- PMID: 20669327
- PMCID: PMC6005705
- DOI: 10.1002/mds.23221
Early-onset L-dopa-responsive parkinsonism with pyramidal signs due to ATP13A2, PLA2G6, FBXO7 and spatacsin mutations
Abstract
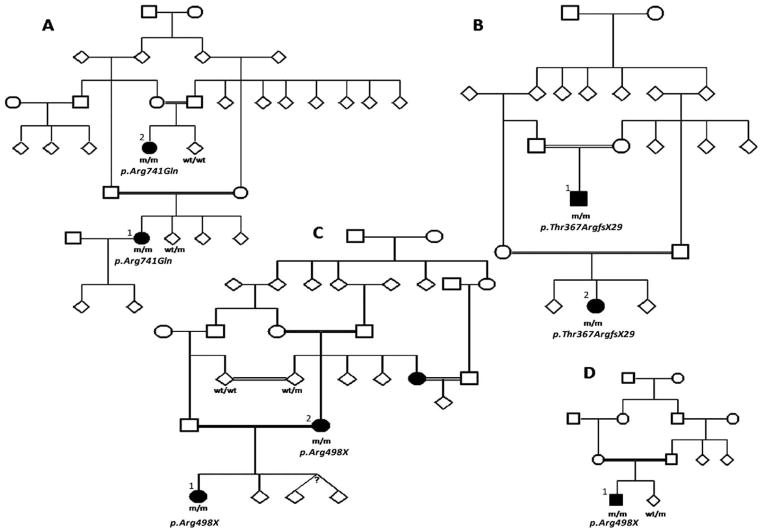
Seven autosomal recessive genes associated with juvenile and young-onset Levodopa-responsive parkinsonism have been identified. Mutations in PRKN, DJ-1, and PINK1 are associated with a rather pure parkinsonian phenotype, and have a more benign course with sustained treatment response and absence of dementia. On the other hand, Kufor-Rakeb syndrome has additional signs, which distinguish it clearly from Parkinson's disease including supranuclear vertical gaze palsy, myoclonic jerks, pyramidal signs, and cognitive impairment. Neurodegeneration with brain iron accumulation type I (Hallervorden-Spatz syndrome) due to mutations in PANK2 gene may share similar features with Kufor-Rakeb syndrome. Mutations in three other genes, PLA2G6 (PARK14), FBXO7 (PARK15), and Spatacsin (SPG11) also produce clinical similar phenotypes in that they presented with rapidly progressive parkinsonism, initially responsive to Levodopa treatment but later, developed additional features including cognitive decline and loss of Levodopa responsiveness. Here, using homozygosity mapping and sequence analysis in families with complex parkinsonisms, we identified genetic defects in the ATP13A2 (1 family), PLA2G6 (1 family) FBXO7 (2 families), and SPG11 (1 family). The genetic heterogeneity was surprising given their initially common clinical features. On careful review, we found the FBXO7 cases to have a phenotype more similar to PRKN gene associated parkinsonism. The ATP13A2 and PLA2G6 cases were more seriously disabled with additional swallowing problems, dystonic features, severe in some, and usually pyramidal involvement including pyramidal weakness. These data suggest that these four genes account for many cases of Levodopa responsive parkinsonism with pyramidal signs cases formerly categorized clinically as pallido-pyramidal syndrome.
© 2010 Movement Disorder Society.
Conflict of interest statement
Potential conflict of interest: Nothing to report.
Figures
Similar articles
-
Early-onset autosomal-recessive parkinsonian-pyramidal syndrome.Acta Neurol Taiwan. 2012 Sep;21(3):99-107. Acta Neurol Taiwan. 2012. PMID: 23196729 Review.
-
Novel ATP13A2 (PARK9) homozygous mutation in a family with marked phenotype variability.Neurogenetics. 2011 Feb;12(1):33-9. doi: 10.1007/s10048-010-0259-0. Epub 2010 Sep 21. Neurogenetics. 2011. PMID: 20853184 Free PMC article.
-
Altered apoptosis regulation in Kufor-Rakeb syndrome patients with mutations in the ATP13A2 gene.J Cell Mol Med. 2012 Aug;16(8):1916-23. doi: 10.1111/j.1582-4934.2011.01488.x. J Cell Mol Med. 2012. PMID: 22117566 Free PMC article.
-
FBXO7 mutations cause autosomal recessive, early-onset parkinsonian-pyramidal syndrome.Neurology. 2009 Jan 20;72(3):240-5. doi: 10.1212/01.wnl.0000338144.10967.2b. Epub 2008 Nov 26. Neurology. 2009. PMID: 19038853
-
Rare causes of dystonia parkinsonism.Curr Neurol Neurosci Rep. 2010 Nov;10(6):431-9. doi: 10.1007/s11910-010-0136-0. Curr Neurol Neurosci Rep. 2010. PMID: 20694531 Review.
Cited by
-
Impaired proteasome activity and neurodegeneration with brain iron accumulation in FBXO7 defect.Ann Clin Transl Neurol. 2020 Aug;7(8):1436-1442. doi: 10.1002/acn3.51095. Epub 2020 Aug 6. Ann Clin Transl Neurol. 2020. PMID: 32767480 Free PMC article.
-
Linking F-box protein 7 and parkin to neuronal degeneration in Parkinson's disease (PD).Mol Brain. 2016 Apr 18;9:41. doi: 10.1186/s13041-016-0218-2. Mol Brain. 2016. PMID: 27090516 Free PMC article. Review.
-
Non-Motor Symptoms in PLA2G6-Associated Dystonia-Parkinsonism: A Case Report and Literature Review.J Clin Med. 2022 Mar 13;11(6):1590. doi: 10.3390/jcm11061590. J Clin Med. 2022. PMID: 35329915 Free PMC article.
-
Neuropathology and pathogenesis of extrapyramidal movement disorders: a critical update-I. Hypokinetic-rigid movement disorders.J Neural Transm (Vienna). 2019 Aug;126(8):933-995. doi: 10.1007/s00702-019-02028-6. Epub 2019 Jun 18. J Neural Transm (Vienna). 2019. PMID: 31214855 Review.
-
The neuropathology of neurodegeneration with brain iron accumulation.Int Rev Neurobiol. 2013;110:165-94. doi: 10.1016/B978-0-12-410502-7.00009-0. Int Rev Neurobiol. 2013. PMID: 24209439 Free PMC article. Review.
References
-
- Davison C. Pallido-pyramidal disease. J Neuropathol Exp Neurol. 1954;13:50–59. - PubMed
-
- Panagariya A, Sharma B, Dev A. Pallido-pyramidal syndrome: a rare entity. Indian J Med Sci. 2007;61:156–157. - PubMed
-
- Tranchant C, Boulay C, Warter JM. Pallido-pyramidal syndrome: an unrecognized entity. Rev Neurol (Paris) 1991;147:308–310. - PubMed
-
- Kitada T, Asakawa S, Hattori N, et al. Mutations in the parkin gene cause autosomal recessive juvenile parkinsonism. Nature. 1998;392:605–608. - PubMed
-
- Ichinose H, Ohye T, Takahashi E, et al. Hereditary progressive dystonia with marked diurnal fluctuation caused by mutations in the GTP cyclohydrolase I gene. Nat Genet. 1994;8:236–242. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
