

MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain
- PMID: 20647238
- PMCID: PMC2928499
- DOI: 10.1101/gr.106849.110
MicroRNA, mRNA, and protein expression link development and aging in human and macaque brain
Abstract
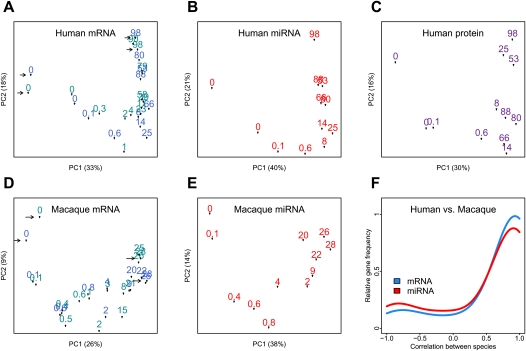
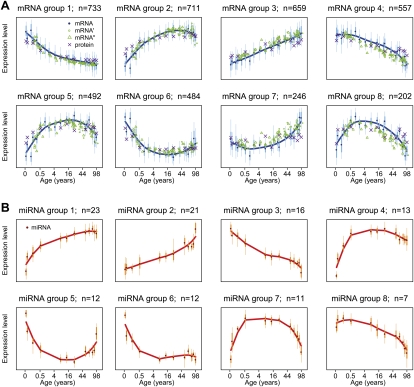
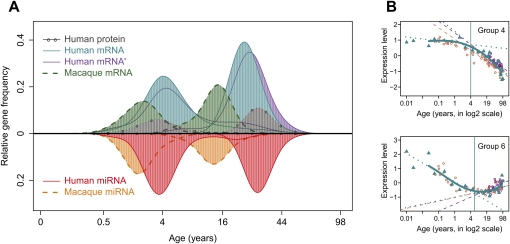
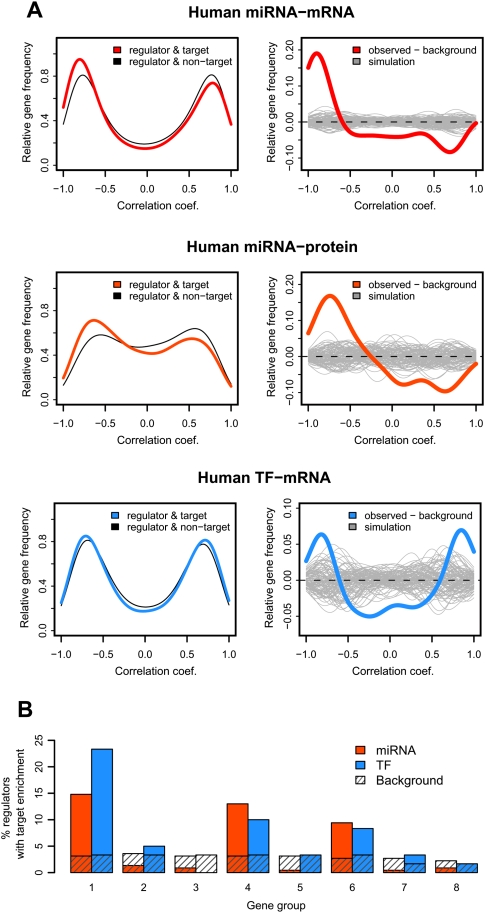
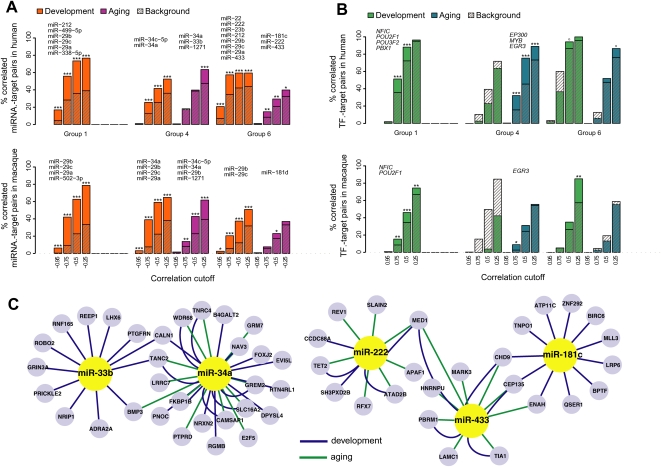
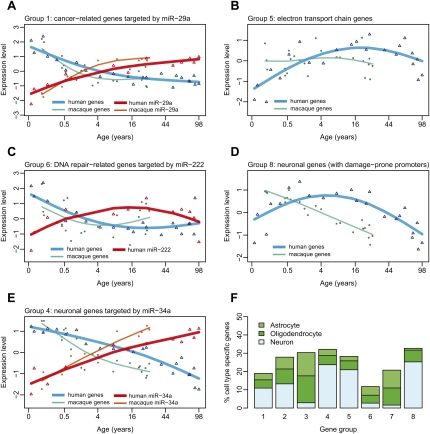
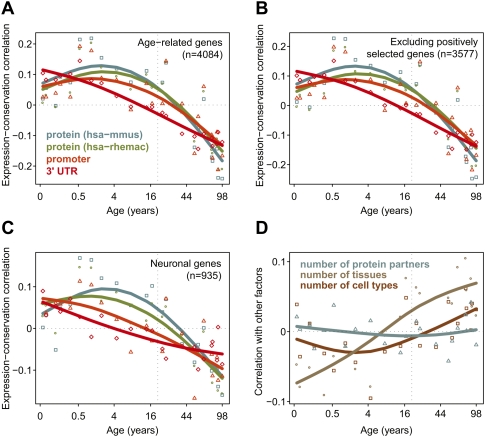
Changes in gene expression levels determine differentiation of tissues involved in development and are associated with functional decline in aging. Although development is tightly regulated, the transition between development and aging, as well as regulation of post-developmental changes, are not well understood. Here, we measured messenger RNA (mRNA), microRNA (miRNA), and protein expression in the prefrontal cortex of humans and rhesus macaques over the species' life spans. We find that few gene expression changes are unique to aging. Instead, the vast majority of miRNA and gene expression changes that occur in aging represent reversals or extensions of developmental patterns. Surprisingly, many gene expression changes previously attributed to aging, such as down-regulation of neural genes, initiate in early childhood. Our results indicate that miRNA and transcription factors regulate not only developmental but also post-developmental expression changes, with a number of regulatory processes continuing throughout the entire life span. Differential evolutionary conservation of the corresponding genomic regions implies that these regulatory processes, although beneficial in development, might be detrimental in aging. These results suggest a direct link between developmental regulation and expression changes taking place in aging.
Figures
Similar articles
-
Transcript and protein expression decoupling reveals RNA binding proteins and miRNAs as potential modulators of human aging.Genome Biol. 2015 Feb 22;16(1):41. doi: 10.1186/s13059-015-0608-2. Genome Biol. 2015. PMID: 25853883 Free PMC article.
-
MicroRNA-driven developmental remodeling in the brain distinguishes humans from other primates.PLoS Biol. 2011 Dec;9(12):e1001214. doi: 10.1371/journal.pbio.1001214. Epub 2011 Dec 6. PLoS Biol. 2011. PMID: 22162950 Free PMC article.
-
MicroRNA expression and regulation in human, chimpanzee, and macaque brains.PLoS Genet. 2011 Oct;7(10):e1002327. doi: 10.1371/journal.pgen.1002327. Epub 2011 Oct 13. PLoS Genet. 2011. PMID: 22022286 Free PMC article.
-
Messenger RNA decay during aging and development.Ageing Res Rev. 2002 Sep;1(4):607-25. doi: 10.1016/s1568-1637(02)00023-5. Ageing Res Rev. 2002. PMID: 12208236 Review.
-
Genomic organization of microRNAs.J Cell Physiol. 2010 Mar;222(3):540-5. doi: 10.1002/jcp.21993. J Cell Physiol. 2010. PMID: 20020507 Free PMC article. Review.
Cited by
-
Systems biology in aging: linking the old and the young.Curr Genomics. 2012 Nov;13(7):558-65. doi: 10.2174/138920212803251418. Curr Genomics. 2012. PMID: 23633915 Free PMC article.
-
Reduced proteasome activity in the aging brain results in ribosome stoichiometry loss and aggregation.Mol Syst Biol. 2020 Jun;16(6):e9596. doi: 10.15252/msb.20209596. Mol Syst Biol. 2020. PMID: 32558274 Free PMC article.
-
Transcript and protein expression decoupling reveals RNA binding proteins and miRNAs as potential modulators of human aging.Genome Biol. 2015 Feb 22;16(1):41. doi: 10.1186/s13059-015-0608-2. Genome Biol. 2015. PMID: 25853883 Free PMC article.
-
MicroRNAs in age-related diseases.EMBO Mol Med. 2013 Feb;5(2):180-90. doi: 10.1002/emmm.201201986. Epub 2013 Jan 22. EMBO Mol Med. 2013. PMID: 23339066 Free PMC article. Review.
-
Identification and characterization of novel and conserved microRNAs in several tissues of the Chinese rare minnow (Gobiocypris rarus) based on illumina deep sequencing technology.BMC Genomics. 2016 Apr 12;17:283. doi: 10.1186/s12864-016-2606-5. BMC Genomics. 2016. PMID: 27066897 Free PMC article.
References
-
- Boehm M, Slack F 2005. A developmental timing microRNA and its target regulate life span in C. elegans. Science 310: 1954–1957 - PubMed
-
- Cahoy JD, Emery B, Kaushal A, Foo LC, Zamanian JL, Christopherson KS, Xing Y, Lubischer JL, Krieg PA, Krupenko SA, et al. 2008. A transcriptome database for astrocytes, neurons, and oligodendrocytes: A new resource for understanding brain development and function. J Neurosci 28: 264–278 - PMC - PubMed
MeSH terms
Substances
Associated data
- Actions
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases