

Bisulfite Patch PCR enables multiplexed sequencing of promoter methylation across cancer samples
- PMID: 20627893
- PMCID: PMC2928506
- DOI: 10.1101/gr.101212.109
Bisulfite Patch PCR enables multiplexed sequencing of promoter methylation across cancer samples
Abstract
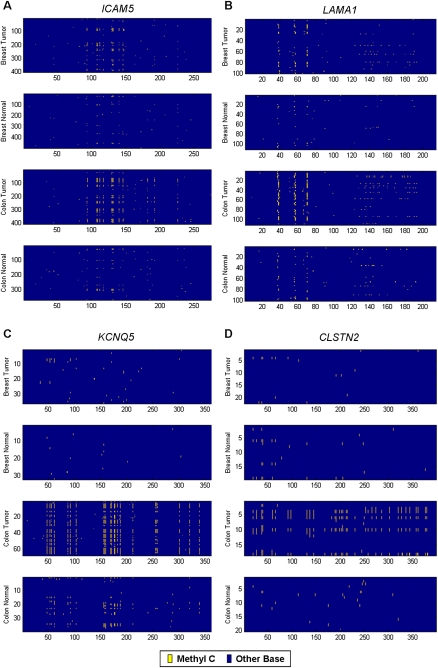
Aberrant DNA methylation frequently occurs at gene promoters during cancer progression. It is important to identify these loci because they are often misregulated and drive tumorigenesis. Bisulfite sequencing is the most direct and highest resolution assay for identifying aberrant promoter methylation. Recently, genomic capture methods have been combined with next-generation sequencing to enable genome-scale surveys of methylation in individual samples. However, it is challenging to validate candidate loci identified by these approaches because an efficient method to bisulfite sequence more than 50 differentially methylated loci across a large number of samples does not exist. To address this problem, we developed Bisulfite Patch PCR, which enables highly multiplexed bisulfite PCR and sequencing across many samples. Using this method, we successfully amplified 100% of 94 targeted gene promoters simultaneously in the same reaction. By incorporating sample-specific DNA barcodes into the amplicons, we analyzed 48 samples in a single run of the 454 Life Sciences (Roche) FLX sequencer. The method requires small amounts of starting DNA (250 ng) and does not require a shotgun library construction. The method was highly specific; 90% of sequencing reads aligned to targeted loci. The targeted promoters were from genes that are frequently mutated in breast and colon cancer, and the samples included breast and colon tumor and adjacent normal tissue. This approach allowed us to identify nine gene promoters that exhibit tumor-specific DNA methylation defects that occur frequently in colon and breast cancer. We also analyzed single nucleotide polymorphisms to observe DNA methylation that accumulated on specific alleles during tumor development. This method is broadly applicable for studying DNA methylation across large numbers of patient samples using next-generation sequencing.
Figures




Similar articles
-
Analyzing the cancer methylome through targeted bisulfite sequencing.Cancer Lett. 2013 Nov 1;340(2):171-8. doi: 10.1016/j.canlet.2012.10.040. Epub 2012 Nov 28. Cancer Lett. 2013. PMID: 23200671 Free PMC article. Review.
-
Application of microdroplet PCR for large-scale targeted bisulfite sequencing.Genome Res. 2011 Oct;21(10):1738-45. doi: 10.1101/gr.116863.110. Epub 2011 Jul 14. Genome Res. 2011. PMID: 21757609 Free PMC article.
-
Quantitative and multiplexed DNA methylation analysis using long-read single-molecule real-time bisulfite sequencing (SMRT-BS).BMC Genomics. 2015 May 6;16(1):350. doi: 10.1186/s12864-015-1572-7. BMC Genomics. 2015. PMID: 25943404 Free PMC article.
-
Microdroplet PCR for Highly Multiplexed Targeted Bisulfite Sequencing.Methods Mol Biol. 2018;1708:333-348. doi: 10.1007/978-1-4939-7481-8_17. Methods Mol Biol. 2018. PMID: 29224152
-
Limitations and advantages of MS-HRM and bisulfite sequencing for single locus methylation studies.Expert Rev Mol Diagn. 2010 Jul;10(5):575-80. doi: 10.1586/erm.10.46. Expert Rev Mol Diagn. 2010. PMID: 20629507 Review.
Cited by
-
Analyzing the cancer methylome through targeted bisulfite sequencing.Cancer Lett. 2013 Nov 1;340(2):171-8. doi: 10.1016/j.canlet.2012.10.040. Epub 2012 Nov 28. Cancer Lett. 2013. PMID: 23200671 Free PMC article. Review.
-
Loss of G0/G1 switch gene 2 (G0S2) promotes disease progression and drug resistance in chronic myeloid leukaemia (CML) by disrupting glycerophospholipid metabolism.Clin Transl Med. 2022 Dec;12(12):e1146. doi: 10.1002/ctm2.1146. Clin Transl Med. 2022. PMID: 36536477 Free PMC article.
-
Analysis of DNA modifications in aging research.Geroscience. 2018 Feb;40(1):11-29. doi: 10.1007/s11357-018-0005-3. Epub 2018 Jan 11. Geroscience. 2018. PMID: 29327208 Free PMC article. Review.
-
Advancing the use of Lactobacillus acidophilus surface layer protein A for the treatment of intestinal disorders in humans.Gut Microbes. 2015;6(6):392-7. doi: 10.1080/19490976.2015.1107697. Gut Microbes. 2015. PMID: 26647142 Free PMC article.
-
Paternal germ line aging: DNA methylation age prediction from human sperm.BMC Genomics. 2018 Oct 22;19(1):763. doi: 10.1186/s12864-018-5153-4. BMC Genomics. 2018. PMID: 30348084 Free PMC article.
References
-
- Baylin SB, Herman JG, Graff JR, Vertino PM, Issa JP 1998. Alterations in DNA methylation: A fundamental aspect of neoplasia. Adv Cancer Res 72: 141–196 - PubMed
-
- Chan TA, Glockner S, Yi JM, Chen W, Van Neste L, Cope L, Herman JG, Velculescu V, Schuebel KE, Ahuja N, et al. 2008. Convergence of mutation and epigenetic alterations identifies common genes in cancer that predict for poor prognosis. PLoS Med 5: e114 doi: 10.1371/journal.pmed.0050114 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources