

Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner
- PMID: 20585398
- PMCID: PMC2886842
- DOI: 10.1371/journal.pone.0011160
Bryostatin modulates latent HIV-1 infection via PKC and AMPK signaling but inhibits acute infection in a receptor independent manner
Abstract
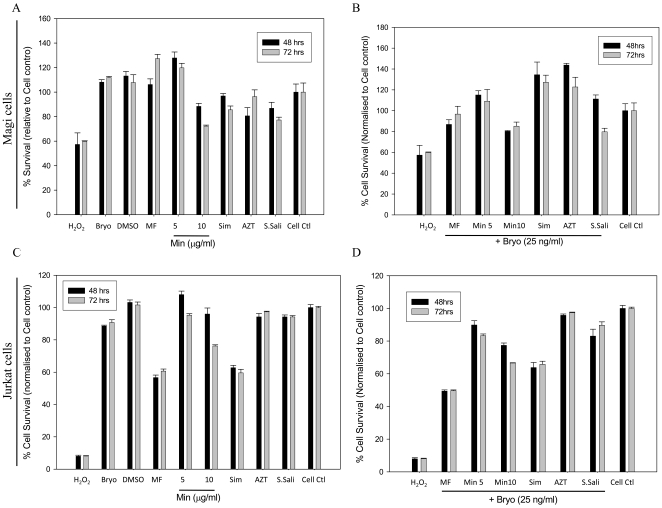
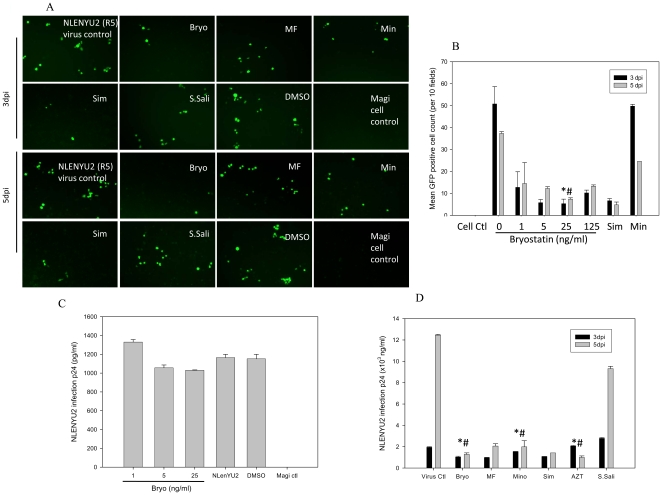
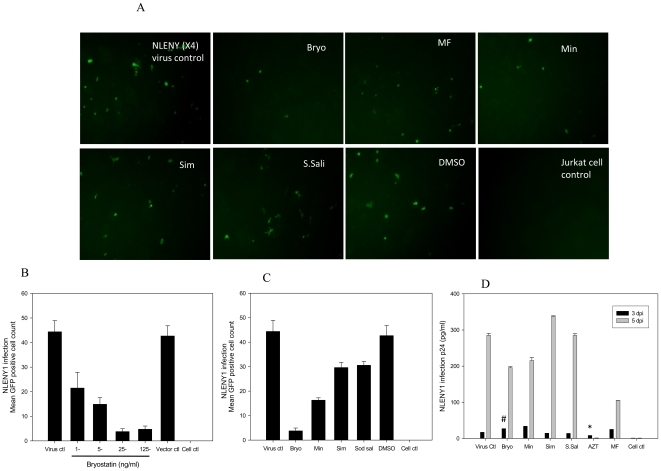
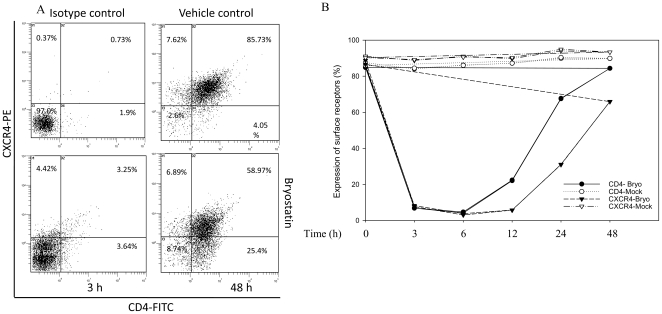
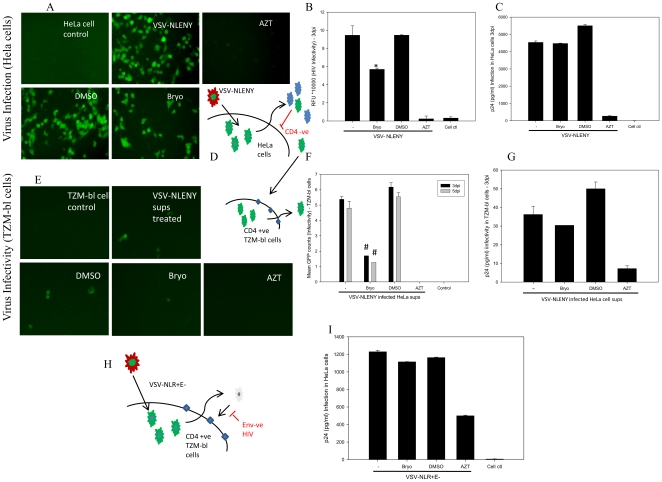
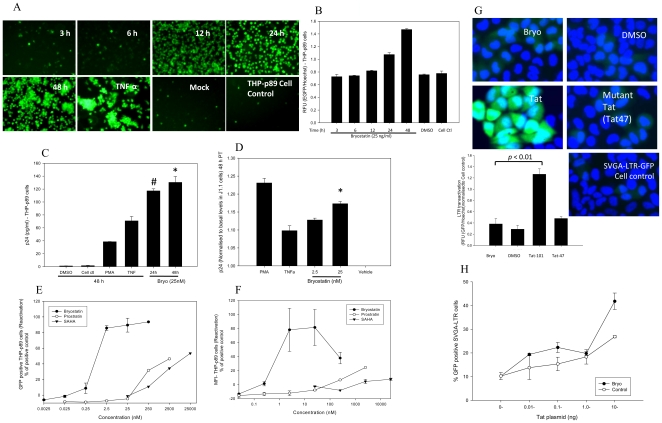
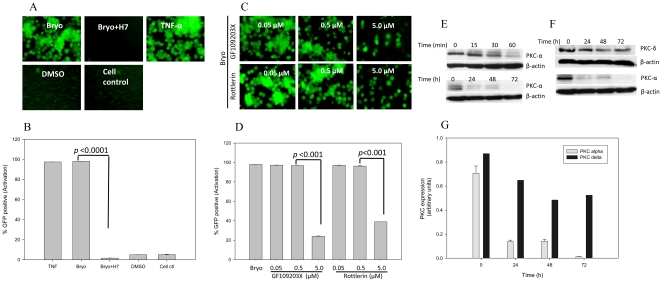
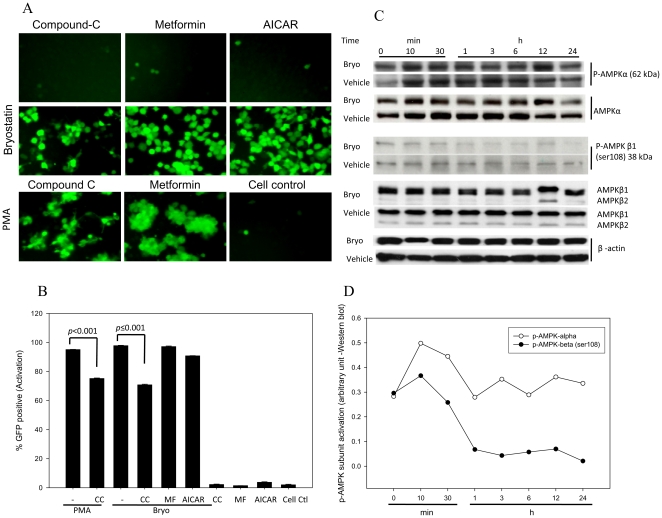
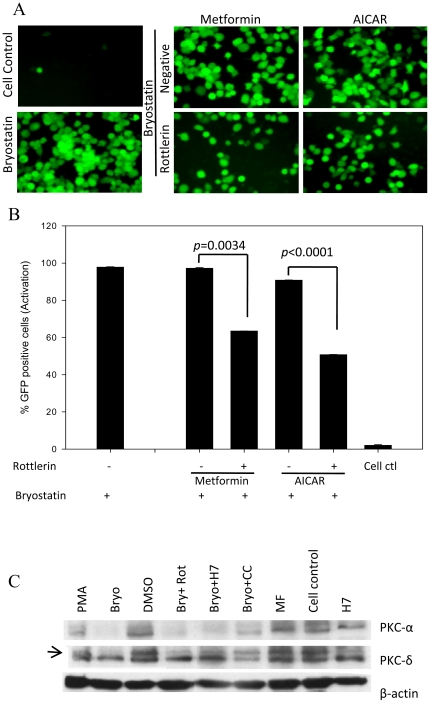
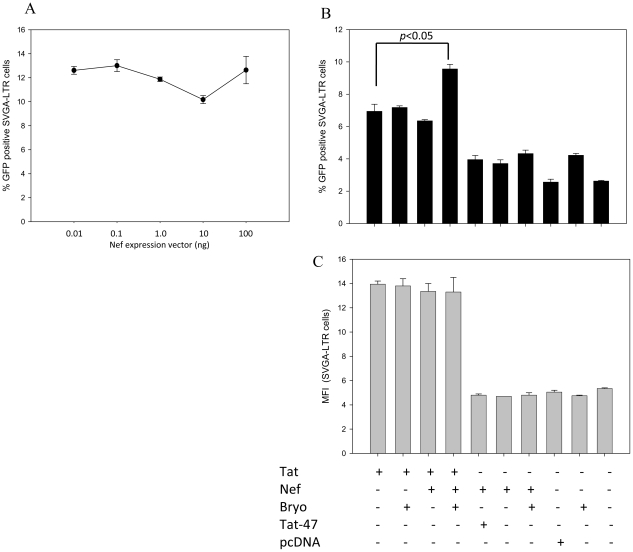
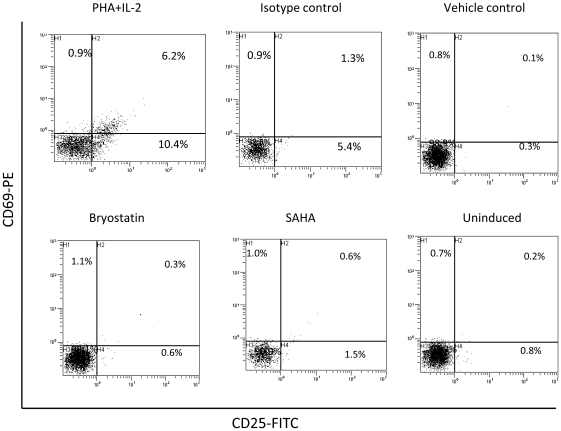
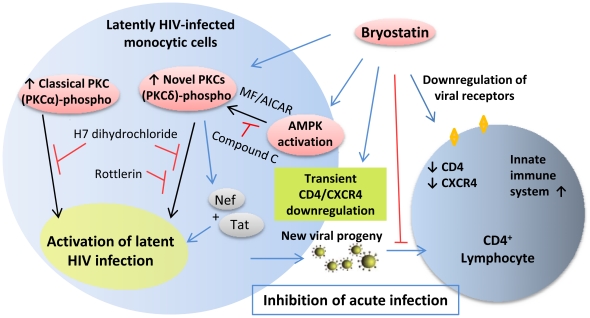
HIV's ability to establish long-lived latent infection is mainly due to transcriptional silencing in resting memory T lymphocytes and other non dividing cells including monocytes. Despite an undetectable viral load in patients treated with potent antiretrovirals, current therapy is unable to purge the virus from these latent reservoirs. In order to broaden the inhibitory range and effectiveness of current antiretrovirals, the potential of bryostatin was investigated as an HIV inhibitor and latent activator. Bryostatin revealed antiviral activity against R5- and X4-tropic viruses in receptor independent and partly via transient decrease in CD4/CXCR4 expression. Further, bryostatin at low nanomolar concentrations robustly reactivated latent viral infection in monocytic and lymphocytic cells via activation of Protein Kinase C (PKC) -alpha and -delta, because PKC inhibitors rottlerin and GF109203X abrogated the bryostatin effect. Bryostatin specifically modulated novel PKC (nPKC) involving stress induced AMP Kinase (AMPK) inasmuch as an inhibitor of AMPK, compound C partially ablated the viral reactivation effect. Above all, bryostatin was non-toxic in vitro and was unable to provoke T-cell activation. The dual role of bryostatin on HIV life cycle may be a beneficial adjunct to the treatment of HIV especially by purging latent virus from different cellular reservoirs such as brain and lymphoid organs.
Conflict of interest statement
Figures
Similar articles
-
Bryostatin activates HIV-1 latent expression in human astrocytes through a PKC and NF-ĸB-dependent mechanism.Sci Rep. 2015 Jul 22;5:12442. doi: 10.1038/srep12442. Sci Rep. 2015. PMID: 26199173 Free PMC article.
-
Bryostatin-1 synergizes with histone deacetylase inhibitors to reactivate HIV-1 from latency.Curr HIV Res. 2010 Sep;8(6):418-29. doi: 10.2174/157016210793499312. Curr HIV Res. 2010. PMID: 20636281
-
4-Deoxyphorbol inhibits HIV-1 infection in synergism with antiretroviral drugs and reactivates viral reservoirs through PKC/MEK activation synergizing with vorinostat.Biochem Pharmacol. 2020 Jul;177:113937. doi: 10.1016/j.bcp.2020.113937. Epub 2020 Mar 26. Biochem Pharmacol. 2020. PMID: 32224142
-
Preclinical and Clinical Studies on Bryostatins, A Class of Marine-Derived Protein Kinase C Modulators: A Mini-Review.Curr Top Med Chem. 2020;20(12):1124-1135. doi: 10.2174/1568026620666200325110444. Curr Top Med Chem. 2020. PMID: 32209043 Review.
-
Activation of latent HIV-1 expression by protein kinase C agonists. A novel therapeutic approach to eradicate HIV-1 reservoirs.Curr Drug Targets. 2011 Mar 1;12(3):348-56. doi: 10.2174/138945011794815266. Curr Drug Targets. 2011. PMID: 20955147 Review.
Cited by
-
Ongoing Clinical Trials of Human Immunodeficiency Virus Latency-Reversing and Immunomodulatory Agents.Open Forum Infect Dis. 2016 Oct 7;3(4):ofw189. doi: 10.1093/ofid/ofw189. eCollection 2016 Oct. Open Forum Infect Dis. 2016. PMID: 27757411 Free PMC article. Review.
-
Insertion Depth Modulates Protein Kinase C-δ-C1b Domain Interactions with Membrane Cholesterol as Revealed by MD Simulations.Int J Mol Sci. 2023 Feb 27;24(5):4598. doi: 10.3390/ijms24054598. Int J Mol Sci. 2023. PMID: 36902029 Free PMC article.
-
Scopoletin Reactivates Latent HIV-1 by Inducing NF-κB Expression without Global T Cell Activation.Int J Mol Sci. 2023 Aug 10;24(16):12649. doi: 10.3390/ijms241612649. Int J Mol Sci. 2023. PMID: 37628826 Free PMC article.
-
How Might We Cure HIV?Curr Infect Dis Rep. 2014 Mar;16(3):392. doi: 10.1007/s11908-014-0392-2. Curr Infect Dis Rep. 2014. PMID: 24562540
-
Macrolides: From Toxins to Therapeutics.Toxins (Basel). 2021 May 12;13(5):347. doi: 10.3390/toxins13050347. Toxins (Basel). 2021. PMID: 34065929 Free PMC article. Review.
References
-
- Geeraert L, Kraus G, Pomerantz J. Hide-and-seek: The challenge of viral persistence in HIV-1 infection. Annu Rev Med. 2008;59:487–450. - PubMed
-
- Chun TW, Finzi D, Margolick J, Chadwick K, Schwartz D, et al. In vivo fate of HIV-1-infected T cells: quantitative analysis of the transition to stable latency. Nat Med. 1995;1:1284–1290. - PubMed
-
- Finzi D, Hermankova M, Pierson T, Carruth LM, Buck C, et al. Identification of a reservoir for HIV-1 in patients on highly active antiretroviral therapy. Science. 1997;278:1295–1300. - PubMed
-
- Wong JK, Hezareh M, Günthard HF, Havlir DV, Ignacio CC, et al. Recovery of replication-competent HIV despite prolonged suppression of plasma viremia. Science. 1997;278:1291–1295. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
