

Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1
- PMID: 20573957
- PMCID: PMC2934684
- DOI: 10.1074/jbc.M109.082883
Neurons efficiently repair glutamate-induced oxidative DNA damage by a process involving CREB-mediated up-regulation of apurinic endonuclease 1
Abstract
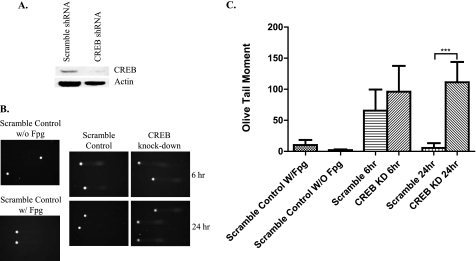
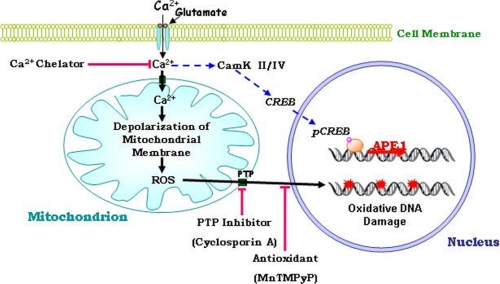
Glutamate, the major excitatory neurotransmitter in the brain, activates receptors coupled to membrane depolarization and Ca(2+) influx that mediates functional responses of neurons including processes such as learning and memory. Here we show that reversible nuclear oxidative DNA damage occurs in cerebral cortical neurons in response to transient glutamate receptor activation using non-toxic physiological levels of glutamate. This DNA damage was prevented by intracellular Ca(2+) chelation, the mitochondrial superoxide dismutase mimetic MnTMPyP (Mn-5,10,15,20-tetra(4-pyridyl)-21H,23H-porphine chloride tetrakis(methochloride)), and blockade of the permeability transition pore. The repair of glutamate-induced DNA damage was associated with increased DNA repair activity and increased mRNA and protein levels of apurinic endonuclease 1 (APE1). APE1 knockdown induced accumulation of oxidative DNA damage after glutamate treatment, suggesting that APE1 is a key repair protein for glutamate-induced DNA damage. A cAMP-response element-binding protein (CREB) binding sequence is present in the Ape1 gene (encodes APE1 protein) promoter and treatment of neurons with a Ca(2+)/calmodulin-dependent kinase inhibitor (KN-93) blocked the ability of glutamate to induce CREB phosphorylation and APE1 expression. Selective depletion of CREB using RNA interference prevented glutamate-induced up-regulation of APE1. Thus, glutamate receptor stimulation triggers Ca(2+)- and mitochondrial reactive oxygen species-mediated DNA damage that is then rapidly repaired by a mechanism involving Ca(2+)-induced, CREB-mediated APE1 expression. Our findings reveal a previously unknown ability of neurons to efficiently repair oxidative DNA lesions after transient activation of glutamate receptors.
Figures






Similar articles
-
BDNF and exercise enhance neuronal DNA repair by stimulating CREB-mediated production of apurinic/apyrimidinic endonuclease 1.Neuromolecular Med. 2014 Mar;16(1):161-174. doi: 10.1007/s12017-013-8270-x. Epub 2013 Oct 10. Neuromolecular Med. 2014. PMID: 24114393 Free PMC article.
-
Oxidative DNA damage is concurrently repaired by base excision repair (BER) and apyrimidinic endonuclease 1 (APE1)-initiated nonhomologous end joining (NHEJ) in cortical neurons.Neuropathol Appl Neurobiol. 2020 Jun;46(4):375-390. doi: 10.1111/nan.12584. Epub 2019 Nov 6. Neuropathol Appl Neurobiol. 2020. PMID: 31628877 Free PMC article.
-
The excitatory neurotransmitter glutamate stimulates DNA repair to increase neuronal resiliency.Mech Ageing Dev. 2011 Aug;132(8-9):405-11. doi: 10.1016/j.mad.2011.06.005. Epub 2011 Jun 25. Mech Ageing Dev. 2011. PMID: 21729715 Free PMC article. Review.
-
Activation of GLP-1 Receptor Enhances Neuronal Base Excision Repair via PI3K-AKT-Induced Expression of Apurinic/Apyrimidinic Endonuclease 1.Theranostics. 2016 Sep 2;6(12):2015-2027. doi: 10.7150/thno.15993. eCollection 2016. Theranostics. 2016. PMID: 27698937 Free PMC article.
-
The DNA base excision repair protein Ape1/Ref-1 as a therapeutic and chemopreventive target.Mol Aspects Med. 2007 Jun-Aug;28(3-4):375-95. doi: 10.1016/j.mam.2007.04.005. Epub 2007 May 3. Mol Aspects Med. 2007. PMID: 17560642 Review.
Cited by
-
Reactive Oxygen Species: Physiological and Physiopathological Effects on Synaptic Plasticity.J Exp Neurosci. 2016 Sep 4;10(Suppl 1):23-48. doi: 10.4137/JEN.S39887. eCollection 2016. J Exp Neurosci. 2016. PMID: 27625575 Free PMC article. Review.
-
Combined exercise and insulin-like growth factor-1 supplementation induces neurogenesis in old rats, but do not attenuate age-associated DNA damage.Rejuvenation Res. 2011 Dec;14(6):585-96. doi: 10.1089/rej.2011.1178. Epub 2011 Aug 25. Rejuvenation Res. 2011. PMID: 21867412 Free PMC article.
-
Induction of chemoresistance by all-trans retinoic acid via a noncanonical signaling in multiple myeloma cells.PLoS One. 2014 Jan 9;9(1):e85571. doi: 10.1371/journal.pone.0085571. eCollection 2014. PLoS One. 2014. PMID: 24416428 Free PMC article.
-
Modulation of DNA base excision repair during neuronal differentiation.Neurobiol Aging. 2013 Jul;34(7):1717-27. doi: 10.1016/j.neurobiolaging.2012.12.016. Epub 2013 Feb 1. Neurobiol Aging. 2013. PMID: 23375654 Free PMC article.
-
DNA repair deficiency in neurodegeneration.Prog Neurobiol. 2011 Jul;94(2):166-200. doi: 10.1016/j.pneurobio.2011.04.013. Epub 2011 Apr 30. Prog Neurobiol. 2011. PMID: 21550379 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous
