

Mutagenesis of adeno-associated virus type 2 capsid protein VP1 uncovers new roles for basic amino acids in trafficking and cell-specific transduction
- PMID: 20573820
- PMCID: PMC2918992
- DOI: 10.1128/JVI.00687-10
Mutagenesis of adeno-associated virus type 2 capsid protein VP1 uncovers new roles for basic amino acids in trafficking and cell-specific transduction
Abstract
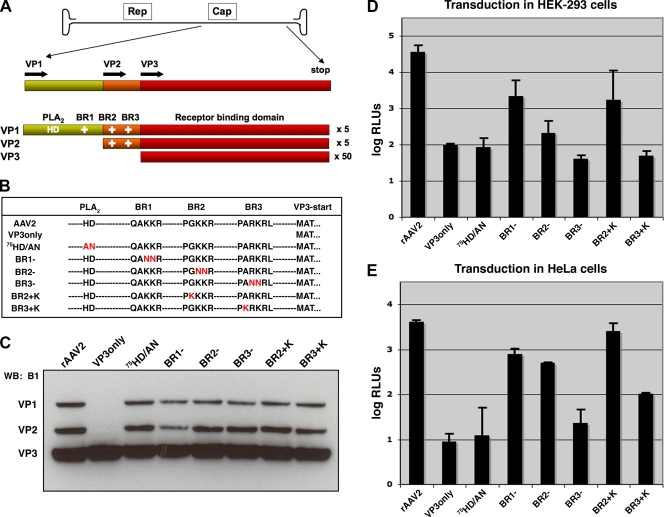
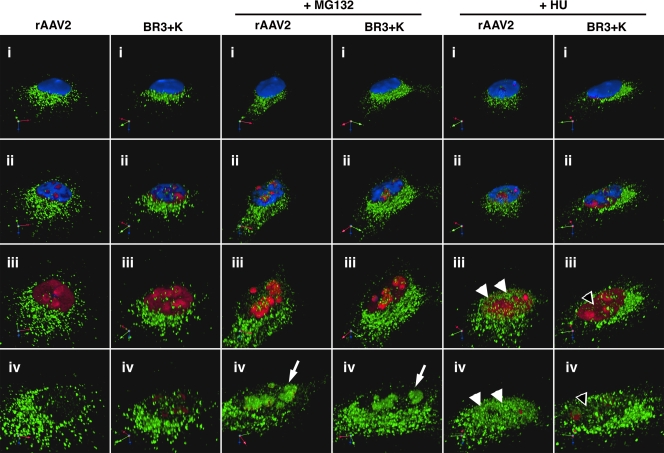
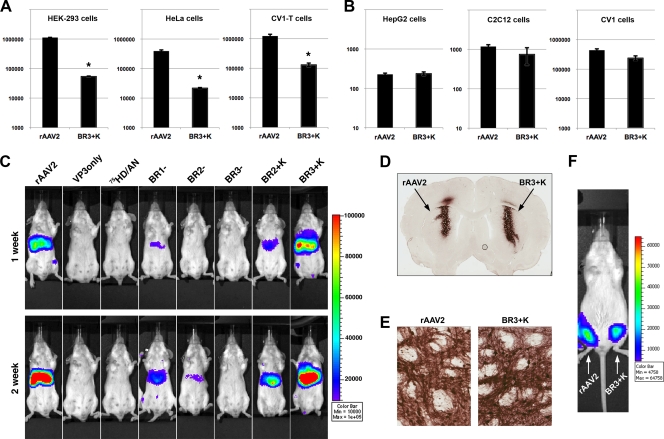
The N termini of the capsid proteins VP1 and VP2 of adeno-associated virus (AAV) play important roles in subcellular steps of infection and contain motifs that are highly homologous to a phospholipase A(2) (PLA(2)) domain and nuclear localization signals (NLSs). To more clearly understand how virion components influence infection, we have generated mutations in these regions and examined their effects on subcellular trafficking, capsid stability, transduction, and sensitivity to pharmacological enhancement. All mutants tested assembled into capsids; retained the correct ratio of VP1, VP2, and VP3; packaged DNA similarly to recombinant AAV2 (rAAV2); and displayed similar stability profiles when heat denatured. Confocal microscopy demonstrated that these mutants trafficked through a perinuclear region in the vicinity of the Golgi apparatus, with a subset of mutants displaying more-diffuse localization consistent with an NLS-deficient phenotype. When tested for viral transduction, two mutant classes emerged. Class I (BR1(-), BR2(-), and BR2+K) displayed partial transduction, whereas class II (VP3 only, (75)HD/AN, BR3(-), and BR3+K) were severely defective. Surprisingly, one class II mutant (BR3+K) trafficked identically to rAAV2 and accumulated in the nucleolus, a step recently described by our laboratory that occurs with wild-type infection. The BR3+K mutant, containing an alanine-to-lysine substitution in the third basic region of VP1, was 10- to 100-fold-less infectious than rAAV2 in transformed cell lines (such as HEK-293, HeLa, and CV1-T cells), but in contrast, it was indistinguishable from rAAV2 in several nontransformed cell lines, as well as in tissues (liver, brain, and muscle) in vivo. Complementation studies with pharmacological adjuvants or adenovirus coinfection suggested that additional positive charges in NLS regions restrict mobilization in the nucleus and limit transduction in a transformed-cell-specific fashion. Remarkably, besides displaying cell-type-specific transduction, this is the first description of a capsid mutant indicating that nuclear entry is not sufficient for AAV-mediated transduction and suggests that additional steps (i.e., subnuclear mobilization or uncoating) limit successful AAV infection.
Figures






Similar articles
-
Nuclear translocation of adeno-associated virus type 2 capsid proteins for virion assembly.J Gen Virol. 2012 Sep;93(Pt 9):1887-1898. doi: 10.1099/vir.0.043232-0. Epub 2012 Jun 13. J Gen Virol. 2012. PMID: 22694902
-
Separate basic region motifs within the adeno-associated virus capsid proteins are essential for infectivity and assembly.J Virol. 2006 Jun;80(11):5199-210. doi: 10.1128/JVI.02723-05. J Virol. 2006. PMID: 16699000 Free PMC article.
-
Identification and characterization of nuclear and nucleolar localization signals in the adeno-associated virus serotype 2 assembly-activating protein.J Virol. 2015 Mar;89(6):3038-48. doi: 10.1128/JVI.03125-14. Epub 2014 Dec 31. J Virol. 2015. PMID: 25552709 Free PMC article.
-
Impact of VP1-specific protein sequence motifs on adeno-associated virus type 2 intracellular trafficking and nuclear entry.J Virol. 2012 Sep;86(17):9163-74. doi: 10.1128/JVI.00282-12. Epub 2012 Jun 13. J Virol. 2012. PMID: 22696661 Free PMC article.
-
The Structures and Functions of Parvovirus Capsids and Missing Pieces: the Viral DNA and Its Packaging, Asymmetrical Features, Nonprotein Components, and Receptor or Antibody Binding and Interactions.J Virol. 2023 Jul 27;97(7):e0016123. doi: 10.1128/jvi.00161-23. Epub 2023 Jun 27. J Virol. 2023. PMID: 37367301 Free PMC article. Review.
Cited by
-
Biophysical and ultrastructural characterization of adeno-associated virus capsid uncoating and genome release.J Virol. 2013 Mar;87(6):2994-3002. doi: 10.1128/JVI.03017-12. Epub 2012 Dec 26. J Virol. 2013. PMID: 23269804 Free PMC article.
-
Adeno-associated virus 2 infection requires endocytosis through the CLIC/GEEC pathway.Cell Host Microbe. 2011 Dec 15;10(6):563-76. doi: 10.1016/j.chom.2011.10.014. Cell Host Microbe. 2011. PMID: 22177561 Free PMC article.
-
The Golgi Calcium ATPase Pump Plays an Essential Role in Adeno-associated Virus Trafficking and Transduction.J Virol. 2020 Oct 14;94(21):e01604-20. doi: 10.1128/JVI.01604-20. Print 2020 Oct 14. J Virol. 2020. PMID: 32817219 Free PMC article.
-
Cytoplasmic trafficking, endosomal escape, and perinuclear accumulation of adeno-associated virus type 2 particles are facilitated by microtubule network.J Virol. 2012 Oct;86(19):10462-73. doi: 10.1128/JVI.00935-12. Epub 2012 Jul 18. J Virol. 2012. PMID: 22811523 Free PMC article.
-
Examining the cross-reactivity and neutralization mechanisms of a panel of mAbs against adeno-associated virus serotypes 1 and 5.J Gen Virol. 2012 Feb;93(Pt 2):347-355. doi: 10.1099/vir.0.035113-0. Epub 2011 Nov 9. J Gen Virol. 2012. PMID: 22071509 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
