

Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate
- PMID: 20566897
- PMCID: PMC3324257
- DOI: 10.1182/blood-2009-11-256354
Overcoming resistance to histone deacetylase inhibitors in human leukemia with the redox modulating compound β-phenylethyl isothiocyanate
Abstract
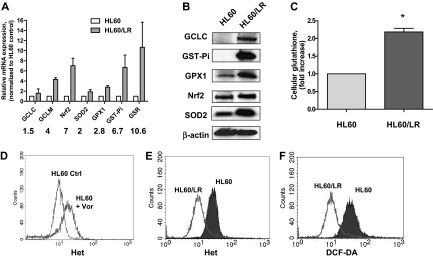
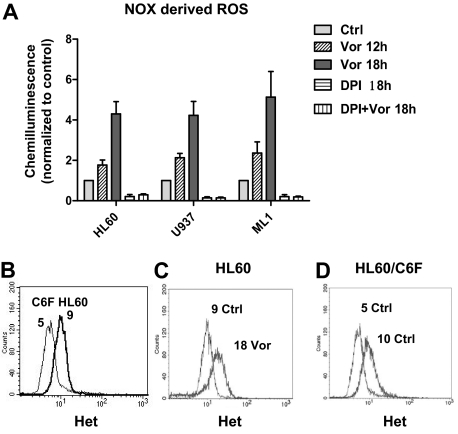
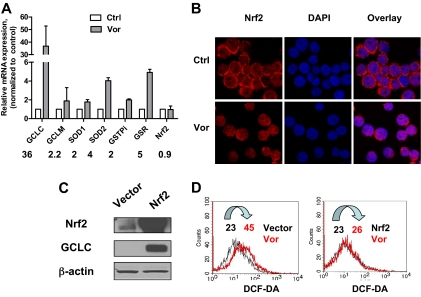
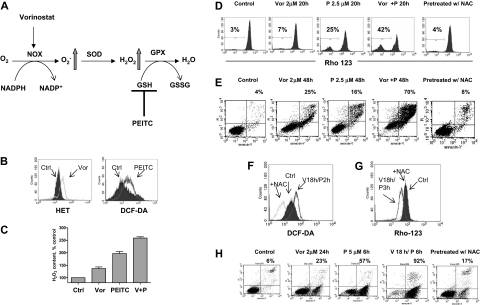
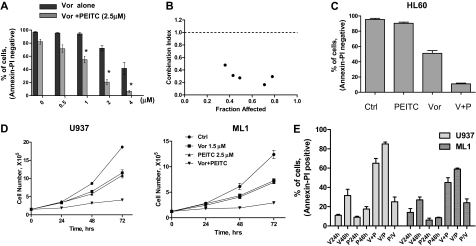
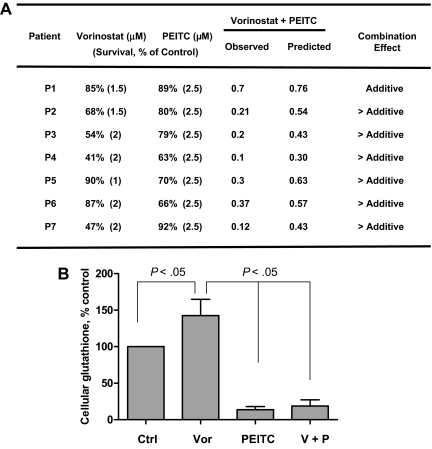
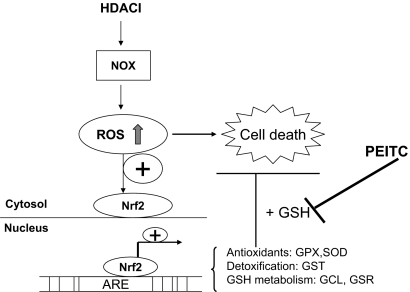
Mechanisms of action and resistance of histone deacetylase inhibitors (HDACIs) are not well understood. A gene expression analysis performed in a phase 1 trial of vorinostat in leukemia indicated that overexpression of genes involved in antioxidant defense was associated with clinical resistance. We hypothesized that nonepigenetic mechanisms may be involved in resistance to HDACI therapy in leukemia. Here we confirmed up-regulation of a series of antioxidants in a pan-HDACI-resistant leukemia cell line HL60/LR. Vorinostat induced reactive oxygen species (ROS) through nicotinamide adenine dinucleotide phosphate oxidase in leukemia cells. An increase in ROS resulted in translocation of nuclear factor E2-related factor 2 from cytosol to nucleus, leading to up-regulation of antioxidant genes, including a majority of glutathione-associated enzymes as a cellular protective mechanism. Addition of β-phenylethyl isothiocyanate, a natural compound capable of depleting cellular glutathione, significantly enhanced the cytotoxicity of vorinostat in leukemia cell lines and primary leukemia cells by inhibiting the cytoprotective antioxidant response. These results suggest that ROS plays an important role in action of vorinostat and that combination with a redox-modulating compound increases sensitivity to HDACIs and also overcomes vorinostat resistance. Such a combination strategy may be an effective therapeutic regimen and have potential clinical application in leukemia.
Figures
Similar articles
-
Resveratrol sensitizes acute myelogenous leukemia cells to histone deacetylase inhibitors through reactive oxygen species-mediated activation of the extrinsic apoptotic pathway.Mol Pharmacol. 2012 Dec;82(6):1030-41. doi: 10.1124/mol.112.079624. Epub 2012 Aug 24. Mol Pharmacol. 2012. PMID: 22923501 Free PMC article.
-
Vorinostat, a histone deacetylase (HDAC) inhibitor, promotes cell cycle arrest and re-sensitizes rituximab- and chemo-resistant lymphoma cells to chemotherapy agents.J Cancer Res Clin Oncol. 2016 Feb;142(2):379-87. doi: 10.1007/s00432-015-2026-y. Epub 2015 Aug 28. J Cancer Res Clin Oncol. 2016. PMID: 26314218
-
Coadministration of histone deacetylase inhibitors and perifosine synergistically induces apoptosis in human leukemia cells through Akt and ERK1/2 inactivation and the generation of ceramide and reactive oxygen species.Cancer Res. 2005 Mar 15;65(6):2422-32. doi: 10.1158/0008-5472.CAN-04-2440. Cancer Res. 2005. PMID: 15781658
-
Redox, cysteines, and kinases-A triad sustaining myeloid leukemia.Adv Cancer Res. 2024;164:1-68. doi: 10.1016/bs.acr.2024.04.008. Epub 2024 May 14. Adv Cancer Res. 2024. PMID: 39306364 Review.
-
Histone deacetylase inhibitors: targeting epigenetic regulation in the treatment of acute leukemia.Ther Adv Hematol. 2024 Oct 16;15:20406207241283277. doi: 10.1177/20406207241283277. eCollection 2024. Ther Adv Hematol. 2024. PMID: 39421716 Free PMC article. Review.
Cited by
-
Synthesis and Antileukemic Activities of Piperlongumine and HDAC Inhibitor Hybrids against Acute Myeloid Leukemia Cells.J Med Chem. 2016 Sep 8;59(17):7974-90. doi: 10.1021/acs.jmedchem.6b00772. Epub 2016 Aug 24. J Med Chem. 2016. PMID: 27505848 Free PMC article.
-
Monitoring Changes in Intracellular Reactive Oxygen Species Levels in Response to Histone Deacetylase Inhibitors.Methods Mol Biol. 2023;2589:337-344. doi: 10.1007/978-1-0716-2788-4_22. Methods Mol Biol. 2023. PMID: 36255635
-
Role of HDACs in normal and malignant hematopoiesis.Mol Cancer. 2020 Jan 7;19(1):5. doi: 10.1186/s12943-019-1127-7. Mol Cancer. 2020. PMID: 31910827 Free PMC article. Review.
-
Improved Anticancer Activities of a New Pentafluorothio-Substituted Vorinostat-Type Histone Deacetylase Inhibitor.Pharmaceuticals (Basel). 2021 Dec 17;14(12):1319. doi: 10.3390/ph14121319. Pharmaceuticals (Basel). 2021. PMID: 34959719 Free PMC article.
-
Valproic Acid Sensitizes Hepatocellular Carcinoma Cells to Proton Therapy by Suppressing NRF2 Activation.Sci Rep. 2017 Nov 8;7(1):14986. doi: 10.1038/s41598-017-15165-3. Sci Rep. 2017. PMID: 29118323 Free PMC article.
References
-
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene. 2007;26(37):5541–5552. - PubMed
-
- Carlisi D, Lauricella M, D'Anneo A, et al. The histone deacetylase inhibitor suberoylanilide hydroxamic acid sensitises human hepatocellular carcinoma cells to TRAIL-induced apoptosis by TRAIL-DISC activation. Eur J Cancer. 2009;45(13):2425–2438. - PubMed
-
- Ruefli AA, Ausserlechner MJ, Bernhard D, et al. The histone deacetylase inhibitor and chemotherapeutic agent suberoylanilide hydroxamic acid (SAHA) induces a cell-death pathway characterized by cleavage of Bid and production of reactive oxygen species. Proc Natl Acad Sci U S A. 2001;98(19):10833–10838. - PMC - PubMed
-
- Peart MJ, Tainton KM, Ruefli AA, et al. Novel mechanisms of apoptosis induced by histone deacetylase inhibitors. Cancer Res. 2003;63(15):4460–4471. - PubMed
-
- Nimmanapalli R, Fuino L, Stobaugh C, Richon V, Bhalla K. Cotreatment with the histone deacetylase inhibitor suberoylanilide hydroxamic acid (SAHA) enhances imatinib-induced apoptosis of Bcr-Abl-positive human acute leukemia cells. Blood. 2003;101(8):3236–3239. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
