

doi: 10.1074/mcp.R110.000067.
Epub 2010 May 27.
Integrative structure modeling of macromolecular assemblies from proteomics data
Affiliations
- PMID: 20507923
- PMCID: PMC2938050
- DOI: 10.1074/mcp.R110.000067
Item in Clipboard
Integrative structure modeling of macromolecular assemblies from proteomics data
Mol Cell Proteomics.
2010 Aug.
Abstract
Proteomics techniques have been used to generate comprehensive lists of protein interactions in a number of species. However, relatively little is known about how these interactions result in functional multiprotein complexes. This gap can be bridged by combining data from proteomics experiments with data from established structure determination techniques. Correspondingly, integrative computational methods are being developed to provide descriptions of protein complexes at varying levels of accuracy and resolution, ranging from complex compositions to detailed atomic structures.
Figures
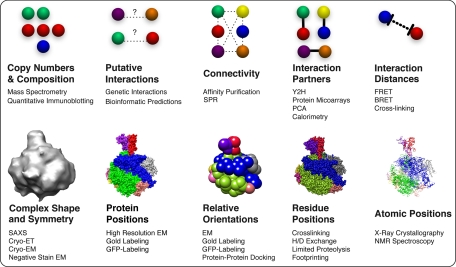
Structural information about a protein assembly. Standard proteomics, biophysical, and computational methods can collectively determine the copy numbers (stoichiometry) and types (composition) of assembly components and predict or experimentally determine protein-protein connectivities (interactivity among a group of proteins) and protein-protein interactions (direct physical interactions). Many of these techniques are capable of a high degree of throughput, allowing for collection of a high volume of data about components of an assembly in a short period of time. Additional biophysical methods can determine distances between components in an assembly, positions of the components, and their relative orientations. Integration of data from varied methods, including low resolution proteomics data, generally increases the accuracy, precision, coverage, and efficiency of structure determination. Methods listed include the following: mass spectrometry (–126), quantitative immunoblotting (127), genetic interactions (128, 129), bioinformatics predictions of protein-protein interactions (130), affinity purification (13, 39, 71, 72), surface plasmon resonance (SPR) (131), Y2H (–116), protein microarrays (–134), protein-fragment complementation assay (PCA) (135, 136), calorimetry (137, 138), FRET (139), bioluminescence resonance energy transfer (BRET) (140), SAXS (24, 25), electron tomography (ET) (21), EM (19, 20, 22), gold labeling (39, 141, 142), green fluorescent protein (GFP) labeling (143), protein-protein docking (144), cross-linking (36, 43, 145, 146), hydrogen/deuterium (H/D) (147), limited proteolysis (148), footprinting (149), x-ray crystallography (15), and NMR spectroscopy (–18).

Determining the molecular architecture of human RNAPII. Top, data gathering. Comparative models of the H-RNAPII subunits were obtained from the ModBase database (54). A density map of H-RNAPII at 20-Å resolution (50) was obtained from the EM data bank (56). Proteomics data for S. cerevisiae RNAPII subunits were obtained from BioGRID (Table III) (55). All pairwise direct interactions are visualized in a single graph with solid edges, and each pulldown experiment is presented as a separate graph with dashed edges to indicate the missing underlying binary interaction network. Pulldowns Rpb1-Rpb2-Rpb3-Rpb4-Rpb5-Rpb8 and Rpb1-Rpb2-Rpb3-Rpb8-Rpb10 are missing some edges for clarity. Gray edges indicate interactions present in BioGRID but not in the yeast RNAPII crystallographic structure. Middle, scoring. The scoring function is the sum of the distance (illustrated between Rpb4 and Rpb7), connectivity (illustrated between Rpb1, Rpb2, Rpb3, Rpb8 and Rpb10), EM quality-of-fit (illustrated between the H-RNAPII density map and Rpb1), and geometric complementarity (illustrated between Rpb4 and Rpb7) restraints. Bottom, optimization. The configuration of the subunits in H-RNAPII was optimized using an extension of the divide-and-conquer MultiFit protocol to incorporate proteomics-based restraints. The optimization procedure resulted in a single model that satisfied all of the input restraints.

Comparison of the crystallographic structure of yeast RNAPII and the integrative model of human RNAPII. I, a–d, atomic representations of the integrative model of H-RNAPII and the reference structure in two views; the reference structure is composed of human subunits individually superposed on their orthologs in the yeast RNAPII structure. The configuration of the H-RNAPII subunits (a and c) is very similar to that in the reference structure (b and d); the Cα RMSD is only 11.4 Å. II, e–h, coarse representations of the H-RNAPII model (e and g) and the reference structure (f and h) in the same two views as in a–d further illustrate the high similarity between the model and the reference. In the coarse representation, sets of 30 contiguous residues are shown as a single bead. III, i and j, protein contact maps for the H-RNAPII model and the reference structure (white, no contact; gray, weak contact; black, contact). The maps are essentially identical, differing only in the interactions of Rpb6 with Rpb2 and Rpb3, and Rpb1 with Rpb12.
Similar articles
-
Structure determination of transient transcription complexes.Biochem Soc Trans. 2016 Aug 15;44(4):1177-82. doi: 10.1042/BST20160079. Biochem Soc Trans. 2016. PMID: 27528766 Review.
-
Modeling of proteins and their assemblies with the integrative modeling platform.Methods Mol Biol. 2011;781:377-97. doi: 10.1007/978-1-61779-276-2_19. Methods Mol Biol. 2011. PMID: 21877292
-
Subunit architecture of intact protein complexes from mass spectrometry and homology modeling.Acc Chem Res. 2008 May;41(5):617-27. doi: 10.1021/ar700218q. Epub 2008 Mar 4. Acc Chem Res. 2008. PMID: 18314965
-
Conserved RNA polymerase II initiation complex structure.Curr Opin Struct Biol. 2017 Dec;47:17-22. doi: 10.1016/j.sbi.2017.03.013. Epub 2017 Apr 22. Curr Opin Struct Biol. 2017. PMID: 28437704 Review.
-
Putting the pieces together: integrative modeling platform software for structure determination of macromolecular assemblies.PLoS Biol. 2012 Jan;10(1):e1001244. doi: 10.1371/journal.pbio.1001244. Epub 2012 Jan 17. PLoS Biol. 2012. PMID: 22272186 Free PMC article.
Cited by
-
UCSF Chimera, MODELLER, and IMP: an integrated modeling system.J Struct Biol. 2012 Sep;179(3):269-78. doi: 10.1016/j.jsb.2011.09.006. Epub 2011 Sep 22. J Struct Biol. 2012. PMID: 21963794 Free PMC article.
-
Molecular architecture and function of the SEA complex, a modulator of the TORC1 pathway.Mol Cell Proteomics. 2014 Nov;13(11):2855-70. doi: 10.1074/mcp.M114.039388. Epub 2014 Jul 29. Mol Cell Proteomics. 2014. PMID: 25073740 Free PMC article.
-
Systems Proteomics View of the Endogenous Human Claudin Protein Family.J Proteome Res. 2016 Feb 5;15(2):339-59. doi: 10.1021/acs.jproteome.5b00769. Epub 2016 Jan 12. J Proteome Res. 2016. PMID: 26680015 Free PMC article. Review.
-
A critical assessment of information-guided protein-protein docking predictions.Mol Cell Proteomics. 2013 Mar;12(3):679-86. doi: 10.1074/mcp.M112.020198. Epub 2012 Dec 13. Mol Cell Proteomics. 2013. PMID: 23242549 Free PMC article.
-
Toward an integrated structural model of the 26S proteasome.Mol Cell Proteomics. 2010 Aug;9(8):1666-77. doi: 10.1074/mcp.R000002-MCP201. Epub 2010 May 13. Mol Cell Proteomics. 2010. PMID: 20467039 Free PMC article.
References
-
- Alberts B. (1998) The cell as a collection of protein machines: preparing the next generation of molecular biologists. Cell 92, 291–294 - PubMed
-
- Abbott A. (2002) Proteomics: the society of proteins. Nature 417, 894–896 - PubMed
-
- Schmeing T. M., Ramakrishnan V. (2009) What recent ribosome structures have revealed about the mechanism of translation. Nature 461, 1234–1242 - PubMed
-
- Allen G. S., Frank J. (2007) Structural insights on the translation initiation complex: ghosts of a universal initiation complex. Mol. Microbiol. 63, 941–950 - PubMed
-
- Horwich A. L., Fenton W. A. (2009) Chaperonin-mediated protein folding: using a central cavity to kinetically assist polypeptide chain folding. Q. Rev. Biophys. 42, 83–116 - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Miscellaneous
