

Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome
- PMID: 20498079
- PMCID: PMC2890780
- DOI: 10.1073/pnas.1000219107
Functional analyses of variants reveal a significant role for dominant negative and common alleles in oligogenic Bardet-Biedl syndrome
Abstract
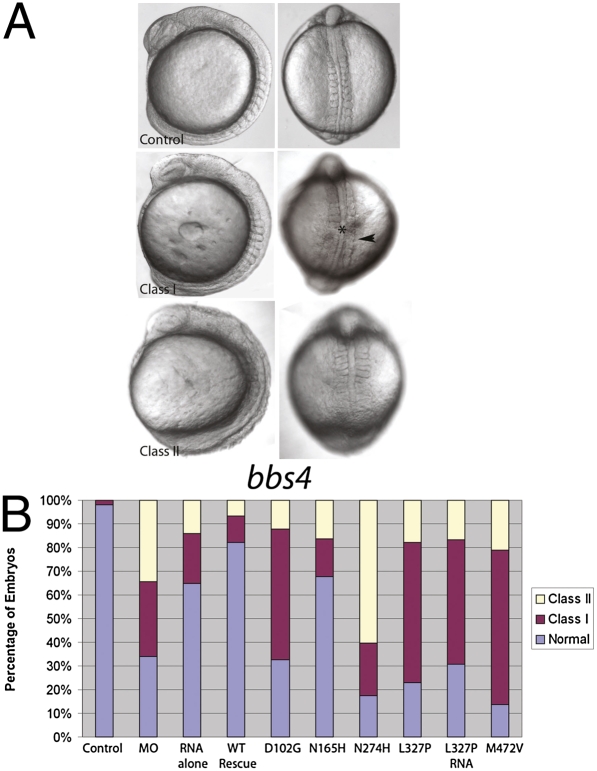
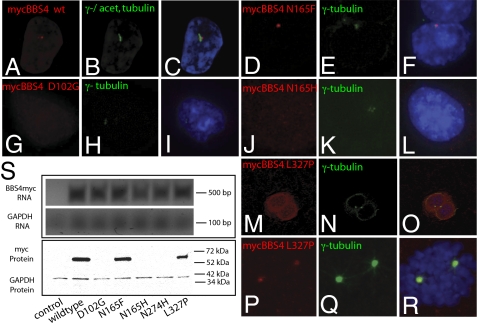
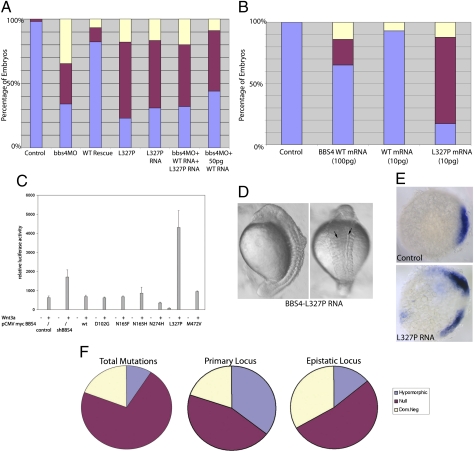
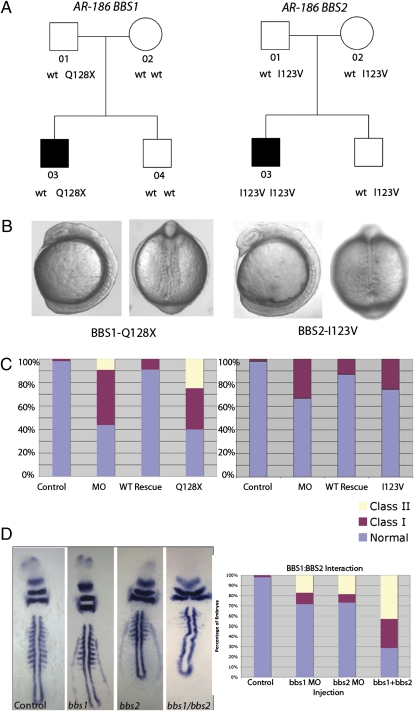
Technological advances hold the promise of rapidly catalyzing the discovery of pathogenic variants for genetic disease. However, this possibility is tempered by limitations in interpreting the functional consequences of genetic variation at candidate loci. Here, we present a systematic approach, grounded on physiologically relevant assays, to evaluate the mutational content (125 alleles) of the 14 genes associated with Bardet-Biedl syndrome (BBS). A combination of in vivo assays with subsequent in vitro validation suggests that a significant fraction of BBS-associated mutations have a dominant-negative mode of action. Moreover, we find that a subset of common alleles, previously considered to be benign, are, in fact, detrimental to protein function and can interact with strong rare alleles to modulate disease presentation. These data represent a comprehensive evaluation of genetic load in a multilocus disease. Importantly, superimposition of these results to human genetics data suggests a previously underappreciated complexity in disease architecture that might be shared among diverse clinical phenotypes.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Comment in
-
Explaining a rare disorder.Lab Anim (NY). 2010 Jul;39(7):196. doi: 10.1038/laban0710-196b. Lab Anim (NY). 2010. PMID: 20567219 No abstract available.
Similar articles
-
Copy-Number Variation Contributes to the Mutational Load of Bardet-Biedl Syndrome.Am J Hum Genet. 2016 Aug 4;99(2):318-36. doi: 10.1016/j.ajhg.2015.04.023. Am J Hum Genet. 2016. PMID: 27486776 Free PMC article.
-
Genetic interaction of BBS1 mutations with alleles at other BBS loci can result in non-Mendelian Bardet-Biedl syndrome.Am J Hum Genet. 2003 May;72(5):1187-99. doi: 10.1086/375178. Epub 2003 Apr 3. Am J Hum Genet. 2003. PMID: 12677556 Free PMC article.
-
The oligogenic properties of Bardet-Biedl syndrome.Hum Mol Genet. 2004 Apr 1;13 Spec No 1:R65-71. doi: 10.1093/hmg/ddh092. Epub 2004 Feb 19. Hum Mol Genet. 2004. PMID: 14976158 Review.
-
Recurrent CNVs and SNVs at the NPHP1 locus contribute pathogenic alleles to Bardet-Biedl syndrome.Am J Hum Genet. 2014 May 1;94(5):745-54. doi: 10.1016/j.ajhg.2014.03.017. Epub 2014 Apr 17. Am J Hum Genet. 2014. PMID: 24746959 Free PMC article.
-
Genetics of human Bardet-Biedl syndrome, an updates.Clin Genet. 2016 Jul;90(1):3-15. doi: 10.1111/cge.12737. Epub 2016 Feb 9. Clin Genet. 2016. PMID: 26762677 Review.
Cited by
-
Exploring Key Challenges of Understanding the Pathogenesis of Kidney Disease in Bardet-Biedl Syndrome.Kidney Int Rep. 2020 Jun 29;5(9):1403-1415. doi: 10.1016/j.ekir.2020.06.017. eCollection 2020 Sep. Kidney Int Rep. 2020. PMID: 32954066 Free PMC article. Review.
-
Zebrafish Models of Neurodevelopmental Disorders: Past, Present, and Future.Front Mol Neurosci. 2018 Aug 29;11:294. doi: 10.3389/fnmol.2018.00294. eCollection 2018. Front Mol Neurosci. 2018. PMID: 30210288 Free PMC article. Review.
-
A novel test for recessive contributions to complex diseases implicates Bardet-Biedl syndrome gene BBS10 in idiopathic type 2 diabetes and obesity.Am J Hum Genet. 2014 Nov 6;95(5):509-20. doi: 10.1016/j.ajhg.2014.09.015. Epub 2014 Oct 16. Am J Hum Genet. 2014. PMID: 25439097 Free PMC article.
-
Where genotype is not predictive of phenotype: towards an understanding of the molecular basis of reduced penetrance in human inherited disease.Hum Genet. 2013 Oct;132(10):1077-130. doi: 10.1007/s00439-013-1331-2. Epub 2013 Jul 3. Hum Genet. 2013. PMID: 23820649 Free PMC article. Review.
-
Gene therapy rescues cilia defects and restores olfactory function in a mammalian ciliopathy model.Nat Med. 2012 Sep;18(9):1423-8. doi: 10.1038/nm.2860. Nat Med. 2012. PMID: 22941275 Free PMC article.
References
-
- Badano JL, et al. Dissection of epistasis in oligogenic Bardet-Biedl syndrome. Nature. 2006;439:326–330. - PubMed
-
- Slavotinek AM, et al. Mutations in MKKS cause Bardet-Biedl syndrome. Nat Genet. 2000;26:15–16. - PubMed
-
- Katsanis N, et al. Mutations in MKKS cause obesity, retinal dystrophy and renal malformations associated with Bardet-Biedl syndrome. Nat Genet. 2000;26:67–70. - PubMed
-
- Mykytyn K, et al. Identification of the gene that, when mutated, causes the human obesity syndrome BBS4. Nat Genet. 2001;28:188–191. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
