

Dominant mutations in the cation channel gene transient receptor potential vanilloid 4 cause an unusual spectrum of neuropathies
- PMID: 20460441
- PMCID: PMC2912694
- DOI: 10.1093/brain/awq109
Dominant mutations in the cation channel gene transient receptor potential vanilloid 4 cause an unusual spectrum of neuropathies
Abstract
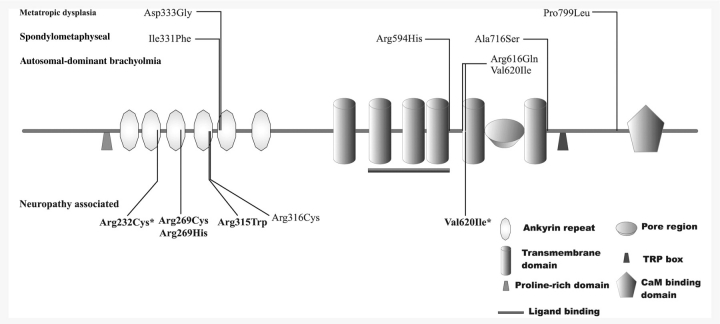
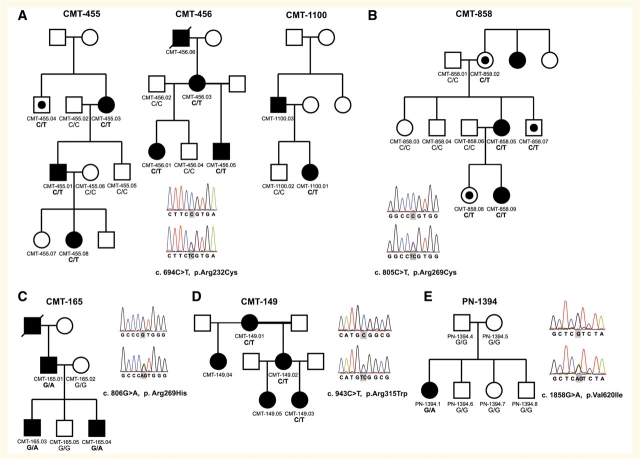
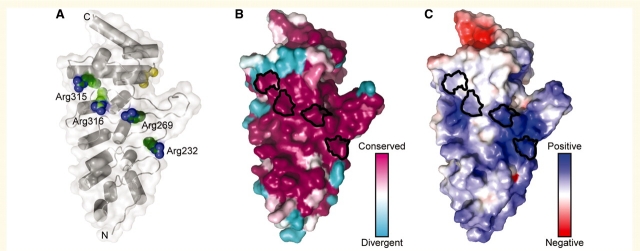
Hereditary neuropathies form a heterogeneous group of disorders for which over 40 causal genes have been identified to date. Recently, dominant mutations in the transient receptor potential vanilloid 4 gene were found to be associated with three distinct neuromuscular phenotypes: hereditary motor and sensory neuropathy 2C, scapuloperoneal spinal muscular atrophy and congenital distal spinal muscular atrophy. Transient receptor potential vanilloid 4 encodes a cation channel previously implicated in several types of dominantly inherited bone dysplasia syndromes. We performed DNA sequencing of the coding regions of transient receptor potential vanilloid 4 in a cohort of 145 patients with various types of hereditary neuropathy and identified five different heterozygous missense mutations in eight unrelated families. One mutation arose de novo in an isolated patient, and the remainder segregated in families. Two of the mutations were recurrent in unrelated families. Four mutations in transient receptor potential vanilloid 4 targeted conserved arginine residues in the ankyrin repeat domain, which is believed to be important in protein-protein interactions. Striking phenotypic variability between and within families was observed. The majority of patients displayed a predominantly, or pure, motor neuropathy with axonal characteristics observed on electrophysiological testing. The age of onset varied widely, ranging from congenital to late adulthood onset. Various combinations of additional features were present in most patients including vocal fold paralysis, scapular weakness, contractures and hearing loss. We identified six asymptomatic mutation carriers, indicating reduced penetrance of the transient receptor potential vanilloid 4 defects. This finding is relatively unusual in the context of hereditary neuropathies and has important implications for diagnostic testing and genetic counselling.
Figures
Similar articles
-
Phenotypic spectrum and incidence of TRPV4 mutations in patients with inherited axonal neuropathy.Neurology. 2014 May 27;82(21):1919-26. doi: 10.1212/WNL.0000000000000450. Epub 2014 Apr 30. Neurology. 2014. PMID: 24789864
-
Phenotypic variability of TRPV4 related neuropathies.Neuromuscul Disord. 2015 Jun;25(6):516-21. doi: 10.1016/j.nmd.2015.03.007. Epub 2015 Mar 18. Neuromuscul Disord. 2015. PMID: 25900305 Free PMC article.
-
TRPV4-pathy manifesting both skeletal dysplasia and peripheral neuropathy: a report of three patients.Am J Med Genet A. 2012 Apr;158A(4):795-802. doi: 10.1002/ajmg.a.35268. Epub 2012 Mar 14. Am J Med Genet A. 2012. PMID: 22419508
-
TRPV4 axonal neuropathy spectrum disorder.J Clin Neurosci. 2012 Jul;19(7):927-33. doi: 10.1016/j.jocn.2011.12.003. Epub 2012 May 20. J Clin Neurosci. 2012. PMID: 22617546 Review.
-
TRPV4-associated skeletal dysplasias.Am J Med Genet C Semin Med Genet. 2012 Aug 15;160C(3):190-204. doi: 10.1002/ajmg.c.31335. Epub 2012 Jul 12. Am J Med Genet C Semin Med Genet. 2012. PMID: 22791502 Review.
Cited by
-
Why individual thermo sensation and pain perception varies? Clue of disruptive mutations in TRPVs from 2504 human genome data.Channels (Austin). 2016 Sep 2;10(5):339-345. doi: 10.1080/19336950.2016.1162365. Epub 2016 Mar 10. Channels (Austin). 2016. PMID: 26962677 Free PMC article.
-
Gain-of-function mutations of TRPV4 acting in endothelial cells drive blood-CNS barrier breakdown and motor neuron degeneration in mice.Sci Transl Med. 2024 May 22;16(748):eadk1358. doi: 10.1126/scitranslmed.adk1358. Epub 2024 May 22. Sci Transl Med. 2024. PMID: 38776392 Free PMC article.
-
Incidence and Clinical Features of TRPV4-Linked Axonal Neuropathies in a USA Cohort of Charcot-Marie-Tooth Disease Type 2.Neuromolecular Med. 2020 Mar;22(1):68-72. doi: 10.1007/s12017-019-08564-4. Epub 2019 Aug 29. Neuromolecular Med. 2020. PMID: 31468327
-
The polymodal ion channel transient receptor potential vanilloid 4 modulates calcium flux, spiking rate, and apoptosis of mouse retinal ganglion cells.J Neurosci. 2011 May 11;31(19):7089-101. doi: 10.1523/JNEUROSCI.0359-11.2011. J Neurosci. 2011. PMID: 21562271 Free PMC article.
-
Mutations in TRPV4 cause an inherited arthropathy of hands and feet.Nat Genet. 2011 Oct 2;43(11):1142-6. doi: 10.1038/ng.945. Nat Genet. 2011. PMID: 21964574
References
-
- Arniges M, Fernandez-Fernandez JM, Albrecht N, Schaefer M, Valverde MA. Human TRPV4 channel splice variants revealed a key role of ankyrin domains in multimerization and trafficking. J Biol Chem. 2006;281:1580–6. - PubMed
-
- Auer-Grumbach M, Schlotter-Weigel B, Lochmuller H, Strobl-Wildemann G, Auer-Grumbach P, Fischer R, et al. Phenotypes of the N88S Berardinelli-Seip congenital lipodystrophy 2 mutation. Ann Neurol. 2005;57:415–24. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
